

Причиной Болезни Фабри (БФ) является низкая активность фермента α-галактозидазы А (α-гал А) и прогрессирующее накопление в лизосомах глоботриаозилцерамида (GL-3) в клетках всего организма. Классическая форма заболевания, встречающаяся у мужчин с активностью фермента α-гал А менее 1% и как правило манифестирует в детском возрасте в виде периодических болевых кризов (боли в конечностях, акропарестезии), кожных появлений (ангиокератомы), нарушений потоотделения (ангидроз, гипогидроз и иногда гипергидроз), характерных нарушений прозрачности роговицы и хрусталика, а также протеинурии.
Постепенное прогрессирование снижения функции почек, вплоть до терминальной стадии почечной недостаточности, происходит в третьем-пятом десятилетии жизни. В среднем возрасте в период коррекции почечной недостаточности начинают развиваться сердечно-сосудистые заболевания, которые являются одной из основных причин смертности этих пациентов. Женщины, имеющие гетерозиготную мутацию, как правило, имеют более мягкие проявления, которые дебютируют в более позднем возрасте, чем у мужчин с классическим фенотипом. При этом, у мужчин с активностью фермента α-гал А выше 1% может быть либо кардиологический клинический вариант, который, обычно, манифестирует в шестом-восьмом десятилетии в виде гипертрофии левого желудочка, недостаточности митрального клапана и/или кардиомиопатии, но без ХПН (хронической почечной недостаточности) или почечным вариантом, связанным с ХПН, но без кожных проявлений и без болевых кризов.
Диагностировать атипичные варианты БФ можно только путем скрининга. Предполагать БФ следует у всех пациентов с гипертрофией миокарда неясной этиологии и инсультом, развивающимся в молодом возрасте. У больных с изолированным поражением почек диагноз может быть заподозрен на основании результатов исследования биоптата почки, в том числе при электронной микроскопии.
Диагностика
У мужчин, наиболее эффективным и надежным методом диагностики БФ является выявление дефицита α-галактозидазы (α-гал А) в плазме, изолированных лейкоцитах и / или культивируемых клетках. У женщин, измерение активности фермента α-гал А является ненадежным т.к. многие женщины-носители имеют нормальную активность фермента α-гал А. GLA является единственным геном, мутации в которых, как известно, вызывают БФ. Почти 100% пораженных мужчин имеют верифицированные мутации гена GLA. Молекулярно-генетическое тестирование является наиболее надежным методом обследования женщин-носителей.
Генетическое консультирование
Верифицированный генетический дефект (мутация) гена GLA позволяет с помощью молекулярно-генетических методов выявить на раннем этапе заболевание у родственников пациента с целью раннего старта ферментзаместительной терапии и предотвращения у них развития сердечнососудистых и почечных осложнений.
БФ характеризуется Х-сцепленным рецессивным типом наследования. При семейных формах, мать пораженного мужчины является облигатным носителем заболевания. В редких случаях пораженный мужчина имеет мутацию, возникшую de novo. Женщины — носители имеют шанс передачи мутации гена GLA равный 50% при каждой беременности. Пораженный мужчина передает мутацию всем своим дочерям. Обследование возможных носителей мутации для определения риска развития заболевания у родственников, а также для проведения пренатальной диагностики возможно, если мутация идентифицирована.
Лечение
Разработана патогенетическая ферментзаместительная терапия рекомбинантными препаратами альфа-галактозидазы А, которая используется с 2001 года. Применяют агалсидазу альфа (Реплагал, Shire Human Genetic Therapies) и агалсидазу бета (Фабразим, Genzyme Corporation), которые получают с помощью линий фибробластов кожи человека и яичников китайских хомячков, соответственно.
Для уменьшения болей и акропарестезии назначается дифенилгидантоин, карбамазепин, габапентин. Ингибиторы АПФ или блокаторы ангиотензиновых рецепторов назначаются для коррекции протеинурии.
При ХПН проводится гемодиализ и/или транплантация почек. Роль ферментзаместительной терапии в профилактике отдаленных почечных, сердечных и неврологических нарушений пока не доказана. Однако, экспертами рекомендуется начало заместительной ферментной терапии как можно раньше всем мужчинам с БФ (в том числе детям и лицам с ХПН и почечной трансплантацией), а также женщинам с значимыми клиническими проявлениями, поскольку они имеют высокий риск в отношении сердечнососудистых, цереброваскулярных и неврологических осложнений.
В России в 2013 году Министерством Здравоохранения РФ разработаны Федеральные клинические рекомендации по диагностике и лечению БФ (Проект 19.01.13) . Список исследований, которые необходимо проводить исходно и в динамике приведен в таблице 1. При этом, при наличии показаний интервалы между исследованиями могут быть сокращены.
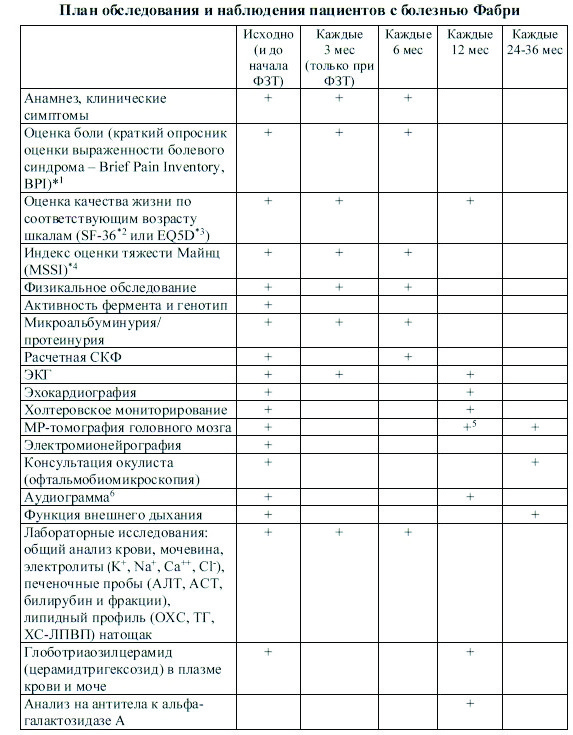 Примечание:
Примечание:
* Используется в Европейских рекомендациях по диагностике и ведению болезни Фабри («Guidelines for the Diagnosis and Management of Anderson-Fabry Disease») под редакторством D.A.Hughes, U.Ramaswam, P.Elliott, P.Deegan, P. Lee, S.Waldek, G. Apperley, T.Cox and A.B.Mehta, 2008;
1 — Создан Pain Research Group, WHO
2 — www.sf-36.org; www.qualitymetric.com;
3 — www.euroqol.org;
4 — Whybra C., Kampmann C., Krummenauer F., Ries M. et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy // Clin. Genet. – 2004. — Apr, №65 (4). – P.299-307.
5 — если была выявлена патология до начала ФЗТ.
6 — если больному больше 5 лет.
Клинический случай:
Мальчик 16 лет, единственный ребенок в семье, родители не состоят в кровнородственном браке, генеалогический анамнез не отягощен. От 1 беременности, протекавшей физиологично, срочных родов. При рождении состояние ребенка удовлетворительное, масса 3450, длина 52см, оценка по шкале АПГАР 7/8 баллов. Раннее развитие по возрасту.
В возрасте 10 лет после физической нагрузки мальчик ощутил боли в ногах, которые купировались самостоятельно. Родители ребенка обратились к ортопеду, патологии выявлено не выявлено. В дальнейшем, после физических нагрузок боли в ногах стали появляться чаще, сопровождались подъемом температуры до фебрильных значений, купировались самостоятельно. Ребенок консультировался в институте ревматологии, где патологии выявлено не было. С целью уточнения диагноза мальчик был госпитализирован в педиатрическое отделение МОНИКИ. Был установлен диагноз «Рецидивирующая инфекция». Учитывая нарастание болевого синдрома (боли участились, появлялись 1-2- раза в месяц), а также присоединились боли в руках, ребенок был обследован в ММА им. Сеченова, где был установлен диагноз «Термоневроз». Назначена седативная и сосудистая терапия. Лечение было без эффекта. В последующие два года боли стали возникать чаще, наросла интенсивность болевых кризов. Боли купировались приемом препарата НАЙЗ. В связи с ухудшением состояния, ребенок был госпитализирован в Детскую психоневрологическую больницу № 18, где была заподозрена болезнь Фабри. В лаборатории наследственных болезней обмена МГНЦ РАМН были проведены энзимодиагностка и ДНК- анализ. Диагноз БФ был подтвержден. После проведенного обследования выявлена умеренная альбуминурия, в динамике без нарастания. Была начата ферментзаместительная терапия препаратом Реплагал. Терапию ребенок переносит хорошо. Спустя 6 месяцев лечения боли купировались. Анальгетические препараты отменены.
Своевременная диагностика и раннее начало фермент заместительной терапии пациентам с БФ позволят предотвратить прогрессирование заболевания и развития серьезных осложнений, существенно влияющих на качество и продолжительность жизни людей с БФ.


