
Игнатьева В. И.1,Моисеев С.В.2, Буланов Н. М.2, Каровайкина Е. А.2, Моисеев А. С.3
1 Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования «Российская академия народного хозяйства и государственной службы при Президенте Российской Федерации» (просп. Вернадского, д. 82, Москва 119571, Россия)
2 Федеральное государственное автономное образовательное учреждение высшего образования Первый Московский государственный медицинский университет имени И. М. Сеченова Министерства здравоохранения Российской Федерации (Сеченовский Университет)
(ул. Россолимо, д. 11/5, Москва 119435, Россия)
3 Федеральное государственное бюджетное образовательное учреждение высшего образования
«Московский государственный университет имени М. В. Ломоносова» (Ленинские горы, д. 1, Москва 119991, Россия)
Резюме
Болезнь Фабри (БФ) – тяжелое орфанное прогрессирующее наследственное заболевание из группы лизосомных болезней накопле- ния, характеризующееся риском развития почечной недостаточности и тяжелых осложнений со стороны сердечно-сосудистой и центральной нервной систем, причиной которого является врожденный дефицит фермента a-галактозидазы А. По имеющимся на настоящий момент сведениям, раннее назначение препаратов ферментозаместительной терапии (ФЗТ) приводит к значительно- му улучшению исходов для пациентов. Цель исследования – оценить число предотвращенных случаев тяжелых осложнений БФ в зависимости от наличия ФЗТ и времени ее начала. Материалы и методы. В модели, построенной на основании опубликованных результатов длительного наблюдения за пациентами с БФ, получающими в качестве ФЗТ агалсидазу альфа (аналогичных данных для агалсидазы бета найдено не было), было рассчитано ожидаемое число случаев развития жизнеугрожающих осложнений в зави- симости от сроков назначения и длительности проведения ФЗТ. Результаты. Проведение ФЗТ у пациентов с БФ в течение пяти лет позволяет сократить на 25% число тяжелых осложнений со стороны сердечно-сосудистой системы и почек. Раннее начало ФЗТ позволяет дополнительно (по сравнению с более поздним началом) предотвратить осложнения более чем в 20% случаев. Заключе- ние. Раннее начало ФЗТ у пациентов с БФ позволяет существенно замедлить развитие у них тяжелых, жизнеугрожающих осложне- ний, увеличить продолжительность и повысить качество их жизни.
Ключевые слова
Болезнь Фабри, лизосомные болезни накопления, редкие заболевания, агалсидаза альфа, агалсидаза бета, ферментозаместитель- ная терапия.
Статья поступила: … 2018 г.; в доработанном виде:….2018 г.; принята к печати:…2018 г.
Конфликт интересов
Авторы заявляют об отсутствии необходимости раскрытия финансовой поддержки или конфликта интересов в отношении данной публикации. Все авторы сделали эквивалентный вклад в подготовку публикации.
Для цитирования
Игнатьева В. И., Моисеев С. В., Буланов Н. М., Каровайкина Е. А., Моисеев А. С., Моделирование влияния ферментозаместительной терапии на развитие жизнеугрожающих исходов у пациентов с болезнью Фабри. ФАРМАКОЭКОНОМИКА. Современная фармакоэкономика и фарма- коэпидемиология. 2018; 11 (4): …
Modeling the effect of enzyme replacement therapy on life-threatening complications in patients with Fabry disease
Ignatyeva V. I.3,Moiseev S.V.2, Bulanov N. M.2, Karovajkina E. A.2, Moiseev A. S.3
1 Russian Presidential Academy of National Economy and Public Administration, Federal State Educational Institution of Higher Professional Education (82 Vernadskogo prospect, Moscow 119571, Russia)
2 I. M. Sechenov First Moscow State Medical University, Ministry of Health of Russia (11/5 Rossolimo Str., Moscow 119435, Russia)
3 Lomonosov Moscow State University (1 Leninskie gory, Moscow 119991, Russia)
Summary
Fabry disease (FD) is a severe lysosome storage disease caused by congenital deficiency of the enzyme a-galactosidase A and characterized by the risk of renal failure combined with cardiovascular and CNS complications. According to the currently available information, the early start of enzyme replacement therapy (ERT) leads to a significant improvement in patient’s condition. The aim of the study is to assess whether the timely ERT prevents severe FD complications and to calculate the number of prevented cases as depending on the time of ERT start. Materials and methods. The proposed model is based on the published results on patients with FD, receiving agalsidase alpha as ERT (no data for agalsidase beta was found). The expected number of cases with life-threatening complications was calculated for different starting time- points and durations of the ERT. Results. In patients with FD, continuous ERT during five years reduces the number of serious cardiovascular and renal complications by 25%. An early start of ERT makes it possible to additionally (as compared with a late start) prevent the complications in more than 20% of cases. Conclusion. The early initiation of RPT in patients with FD can significantly reduce the occurrence of severe life- threatening complications, increase the patients’ survival and improve their quality of life.
Keywords
Fabry disease, lysosomal storage diseases, rare diseases, agalsidase alpha, agalsidase beta, enzyme replacement therapy.
Received: …2018; in the revised form: ….2018; accepted:….2018. Conflict of interests
The authors declare they have nothing to disclosure regarding the funding or conflict of interests with respect to this manuscript.
All authors contributed equally to this article.
For citation
Ignatyeva V. I., Moiseev S. V., Bulanov N. M., Karovajkina E. A., Moiseev A. S. Modeling the effect of enzyme replacement therapy on life-threatening complicationsinpatientswith Fabrydisease. FARMAKOEKONOMIKA. Modernpharmacoeconomicsandpharmacoepidemiology.[FARMAkoEkoNoMIkA. Sovremennaya farmakoekonomika i farmakoepidemiologiya]. 2018; 11 (4): … (in Russian). DOI:
Corresponding author
Address: 11/5 Rossolimo Str., Moscow 119435, Russia. E-mail address: ignateva@hta-rus.ru (Ignatyeva V. I.).
Введение
Болезнь Фабри (БФ), или болезнь Андерсона-Фабри, – тяжелое прогрессирующее наследственное заболевание из группы лизосо- мных болезней накопления, причиной которого является врожден- ный дефицит фермента a-галактозидазы А (a-gal A) [1,2]. Насле- дование БФ сцеплено с Х-хромосомой, в связи с чем, как правило, проявления БФ более выражены и чаще встречаются у мужчин.
Дефицит фермента a-gal A приводит к прогрессирующему от- ложению в лизосомах клеток органов и тканей метаболита гло- ботриаозилцерамида (Gb-3). Он накапливается в эндотелиальных, периваскулярных и гладкомышечных клетках кровеносных сосу- дов, ганглиоцитах вегетативной нервной системы, эпителиальных клетках почечных клубочков и канальцев, кардиомиоцитах, а так- же в роговице, гистиоцитарных и ретикулярных клетках соедини- тельной ткани [3,4].
Первые клинические признаки БФ обычно появляются в под- ростковом и юношеском возрасте, хотя иногда симптомы заболе- вания могут дебютировать в 3-4 года или, наоборот, отсутствуют вплоть до 3-го десятилетия жизни [5-7]. Чаще всего полная кли- ническая картина наблюдается у взрослых пациентов [3,4]. По данным международных клинических исследований, в среднем от дебюта до установления диагноза проходит около 12 лет [8].
Ранняя диагностика данного заболевания осложнена из-за поли- морфной клинической картины, неспецифичности многих началь- ных симптомов, а также малой осведомленности медицинских работников о данном заболевании [3,4]. Частым симптомом явля- ется нейропатическая боль, частота встречаемости которой дости- гает 76% у мужчин и 64% у женщин с болезнью Фабри [9].
По мере прогрессирования основного заболевания происходят необратимые изменения органов-мишеней, прежде всего почек с формированием хронической болезни почек, требующей гемо- диализа и почечной трансплантации; поражение сердца с разви- тием гипертрофии миокарда; поражение центральной нервной системы [5-7,10]. Возникающие на фоне основного заболевания жизнеугрожающие осложнения существенно ухудшают качество жизни и значительно сокращают ее продолжительность, медиана которой у пациентов с БФ менее 60 лет [10]. Выделяют две формы болезни – классическую и атипичную (позднее начало, изолиро- ванное поражение головного мозга, сердца или почек) [11].
Таким образом, БФ является заболеванием, затрагивающим со- циально-значимую группу пациентов трудоспособного возраста. Жизнеугрожающие осложнения БФ, такие как почечная недоста- точность, болезни сердца или инсульт, приводят к преждевремен- ной смерти пациентов [10]. Ухудшение качества жизни вызвано не только последствиями поражения органов-мишеней, но и спек- тром других симптомов: хроническая боль в конечностях, наруше- ние функций желудочно-кишечного тракта, кожи, слуха, депрессия [10,12].
По данным проведенных в различных странах исследований, распространенность БФ составляет от 1:117 000 до 1:476 000 [3]. То есть, можно ожидать, что в Российской Федерации (РФ) при численности населения 146,8 млн человек популяция пациентов с БФ составляет 308-1 254 человека. Вместе с этим количество па- циентов с установленным диагнозом БФ значительно ниже рас- четных цифр, и в 2018 г. в РФ зарегистрировано только порядка 200 пациентов.
В клинике им. Е. М. Тареева за последние 5 лет было обследовано 98 взрослых пациентов с болезнью Фабри. Средний возраст составил 39,8 лет. У большинства пациентов (87,7%) наблюдался классический фенотип заболевания, характеризовавшийся нейро- патической болью (79,6%), ангиокератомами (43,9%), вихревид- ной кератопатией (53,1%). У 82% пациентов имелись признаки поражения почек, в т.ч. у 31,6% – хроническая терминальная по- чечная недостаточность, у 57,1% – гипертрофия миокарда, у 47,9% – очаги в головном мозге, выявленные при магнитно-ре- зонансной томографии. У всех пациентов диагноз был установлен поздно, а средний срок от появления первого симптома до уста- новки диагноза составил 19,0 лет.
В России БФ включена в перечень жизнеугрожающих и хрони- ческих прогрессирующих редких (орфанных) заболеваний, приво- дящих к сокращению продолжительности жизни граждан или их инвалидности перечень «24 нозологии». Лекарственное обеспече- ние пациентов с заболеваниями, включенными в данный пере- чень, сопряжено с рядом обязательств, возложенных на субъекты РФ. Одним из них является ведение региональных сегментов Фе- дерального регистра пациентов (согласно Постановлению Прави- тельства от 26 апреля 2012 г. № 403), являющегося основой для закупок лекарственного обеспечения. Также Постановлением Пра- вительства РФ от 08.12.2017 г. N 1492 «О Программе государ- ственных гарантий бесплатного оказания гражданам медицинской помощи на 2018 год и на плановый период 2019 и 2020 годов» и Федеральным законом от 21.11.2011 N 323-ФЗ «Об основах ох- раны здоровья граждан в РФ» зафиксировано обязательство ре- гиональных Министерств Здравоохранения обеспечивать данную группу пациентов за счет бюджетных ассигнований бюджетов субъектов РФ. Вместе с этим, согласно Определению Конституци- онного Суда РФ от 02.07.2013 N 1054-О, «в случае недостаточно- сти собственных средств для покрытия расходов на обеспечение лекарственными препаратами для лечения заболеваний, включен- ных в Перечень …, субъекты Российской Федерации вправе рас- считывать на оказание им Российской Федерацией в той или иной форме финансовой помощи целевого характера».
С 2001 г. в медицинской практике для лечения БФ используют специфическую патогенетическую ферментозаместительную те- рапию (ФЗТ), которая на настоящий момент считается наиболее эффективным методом лечения БФ. В РФ зарегистрированы два рекомбинантных ферментных препарата ФЗТ для лечения паци- ентов с БФ:
- агалсидаза альфа (Реплагал®, Shire), полученный с исполь- зованием культивированных фибробластов кожи человека;
- агалсидаза бета (Фабразим®, Genzyme), произведенный с по- мощью технологии рекомбинантной ДНК с использованием кле- ток китайского хомячка.
Оба препарата для ФЗТ согласно инструкциям по медицинско- му применению предназначены для регулярного пожизненного внутривенного введения. Агалсидаза альфа вводится внутривенно
15 мг/ч, после установления толерантности она может быть повы- шена.
В настоящий момент оба препарата включены в перечень ЖНВЛП и являются единственной зарегистрированной патогене- тической терапией для БФ в РФ. Вместе с этим ФЗТ в РФ получа- ют менее 50% пациентов с диагностированной БФ, при этом боль- шинство из них находится на терапии агалсидазой альфа.
Данные по клинической эффективности и безопасности препа- ратов ФЗТ, предназначенных для лечения пациентов с БФ, были обобщены в систематизированном обзоре Cochrane [13], вклю- чавшем семь клинических исследований [14-20]. Авторы данного обзора пришли к выводу, что ФЗТ по сравнению с плацебо стати- стически значимо влияет на эндотелиальные депозиты глоботриа- озилцерамида, что подтверждает возможность влияния ФЗТ на последующее развитие жизнеугрожающих осложнений, а так- же предполагает большую эффективность ФЗТ в случае более раннего ее начала. Помимо этого, было продемонстрировано улучшение качества жизни пациентов за счет уменьшения боли. Каких-либо данных, которые указывали бы на статистически зна- чимое превосходство одного из препаратов для ФЗТ, получено не было.
В силу ограничений дизайна РКИ, в частности их краткосрочно- сти, для подтверждения влияния ФЗТ на развитие осложнений и выживаемость пациентов необходимо проведение длительных когортных исследований, в т.ч. на основании регистров. В рамках одного из таких исследований (289 взрослых пациентов и 22 паци- ента детского возраста с БФ) было показано статистически значи- мое положительное влияние длительного приема препаратов ФЗТ на снижение уровня гипертрофии левого желудочка (оценивалась на основании изменения индекса массы левого желудочка) и улуч- шение почечной функции (оценивалась на основании изменения скорости клубочковой фильтрации и риска протеинурии) [21]. Статистически значимых различий между группами пациентов, получавших агалсидазу альфа и агалсидазу бета, выявлено не было. В исследовании, сравнивавшем результаты 5-летнего ле- чения агалсидазой альфа 740 пациентов1 с опубликованными дан- ными для пациентов, не получающих ФЗТ, было показано замед- ление ухудшения функции почек и развития гипертрофии левого желудочка у пациентов, получавших терапию. Также для этих па- циентов был ниже риск развития жизнеугрожающих осложнений, а продолжительность жизни значительно выше (медиана 77,5 лет у мужчин, получавших терапию, по сравнению с 60 годами у не по- лучавших терапию) [22] (рис. 1). Еще один анализ базы данных FOS показал, что раннее начало ФЗТ агалсидазой альфа после манифестации симптомов БФ (менее 24 мес.) было связано со статистически значимо меньшей частотой неблагоприятных со- бытий со стороны сердечно-сосудистой системы и почек по срав- нению с отложенным началом ФЗТ (24 мес. и более после мани- фестации симптомов БФ): относительный риск (ОР) – 0,48 (95% доверительный интервал (ДИ) 0,38-0,70) и ОР 0,54 (95% ДИ 0,37-0,79) соответственно [23] (рис. 2-3).
Согласно «Федеральным клиническим рекомендациям по ди- агностике и лечению болезни Фабри» мужчинам с БФ замести- тельная терапия показана сразу после установления диагноза, а у женщин показаниями к лечению считают выраженные сим- птомы или признаки прогрессирующего поражения органов-ми- шеней, в т.ч. хроническая невропатическая боль в кистях и стопах, резистентная к стандартной терапии [11]. Однако фер- ментозаместительная терапия в настоящее время проводится только у трети больных с БФ. Особенностью терапии БФ в РФ является то, что значительная часть пациентов диагностируется на поздних стадиях от начала заболевания, а начало терапии некапельно в течение 40 мин. в дозе 0,2 мг/кг массы тела 1 раз в 2 недели. Агалсидаза бета также вводится 1 раз в 2 недели, но в дозе 1 мг/кг и начальная скорость инфузии не должна превышать редко откладывается еще на несколько лет в зависимости от ре- гиона, что значительно ухудшает прогноз течения заболевания и результаты лечения.
1 Формирование базы данных «Изучение исходов болезни Фабри» (Fabri Outcome Survey, FOS), из которой извлекались данные, под- держивается компанией Шайер.

Учитывая, что в настоящий момент в РФ имеется выраженная тенденция к позднему началу или отказу от проведения ФЗТ у па- циентов с БФ, целью настоящего исследования была оценка числа предотвратимых в результате проведения ФЗТ случаев жизнеу- грожающих осложнений.
Материалы и методы
Нами была построена математическая модель, позволяющая рассчитать число пациентов, у которых происходят неблагоприят- ные события со стороны сердечно-сосудистой системы и почек, в зависимости от срока назначения препаратов ФЗТ после уста- новления диагноза БФ и длительности терапии (наблюдения). Для построения модели и использовалась программа EXCEL из пакета Microsoft Office (Microsoft, США) и среда статистических вычисле- ний R (факультет статистики университета Мельбурна, Австра- лия).
В основе модели лежат опубликованные данные о частоте раз- вития осложнений со стороны сердечно-сосудистой системы и почек в зависимости от длительности ФЗТ у пациентов с ран- ним и отложенным началом терапии агалсидазой альфа после установления диагноза БФ [23]. Дополнительно нами была рас- считана вероятность наступления комбинированного исхода (смерть, инсульт, осложнения со стороны сердечно-сосудистой системы или почек) для пациентов с БФ, получающих и не полу- чающих агалсидазу альфа, также на основании опубликованных результатов долгосрочного наблюдательного исследования [22]. Аналогичных опубликованных данных для агалсидазы бета най- дено не было.
Для построения модели на основании оцифрованных опублико- ванных кривых выживаемости (до времени наступления изучае- мого события) в среде статистических вычислений R были рекон- струированы индивидуальные данные для пациентов с БФ и построены параметрические модели функций выживаемости [24,25]. Соответствие полученных моделей выживаемости кри- вым выживаемости, полученным в исследованиях, можно оценить по представленным графикам (см. рис. 1-3). Методика расчетов приведена в формулах 1-11.
Конкретные виды распределения для каждого изучаемого исхо- да были выбраны на основании наличия статистической значимо- сти параметров распределения (табл. 1).
Для расчета вероятности сердечно-сосудистых и почечных ос- ложнений у пациентов с БФ при раннем начале ФЗТ была исполь- зована формула (1):
SurvFunc = Exp (–l*t g)
где SurvFunc – это вероятность нахождения в состоянии; t – время;
l – параметр, определяющий масштаб распределения, рассчиты- вающийся по формуле (2):
l = exp (-scale*shape),
где l – параметр, определяющий форму распределения, рассчи- тывающийся по формуле (3):
l = scale.
Вероятность осложнений со стороны сердечно-сосудистой си- стемы у пациентов с БФ при отложенном начале ФЗТ была рассчи- тана по формуле (4):
SurvFunc = 1 – НормРаспр ( (log(t) – μ) / s),
где SurvFunc – это вероятность нахождения в состоянии; t – время; μ – параметр, определяющий масштаб распределения, рассчиты- вающийся по формуле (5):
l = shape,
где s – параметр, определяющий форму распределения, рассчи- тывающийся по формуле (6):
s = scale.
Расчет вероятности осложнений со стороны почек у пациентов с БФ при отложенном начале ФЗТ проводился по формуле (7):
SurvFunc = 1 – НормРаспр ( (log(t) – l) / g),
где t – время; l – параметр, определяющий масштаб логнормаль- ного распределения, рассчитывающийся по формуле (8):
l = shape,
где g – параметр, определяющий форму распределения, рассчи- тывающийся по формуле (9):
l = scale.
Расчет вероятности комбинированных исходов у пациентов с БФ, получающих и не получающих ФЗТ, проводился по формуле (10):
SurvFunc = exp (-l*t),
где t – время; l – параметр, определяющий масштаб логнормаль- ного распределения, рассчитывающийся по формуле (11):
l = exp(–shape).
При моделировании выживаемости без комбинированного ис- хода у пациентов с БФ, получающих ФЗТ, по сравнению с пациен- тами, не получающими терапию, был выбран несколько отличаю- щийся подход. Выживаемость без изучаемого исхода для группы пациентов, не получающих ФЗТ, была смоделирована на основа- нии кривой выживаемости так, как было описано выше. Исходно эти данные были получены для группы пациентов, получавших плацебо в исследовании Banikazemi, 87% в группе составляли мужчины [16]. В когорте FOS (получавшие ФЗТ) доля мужчин
 Рисунок 1. Вероятность отсутствия комбинированного исхода в зависимости от давности наблюдения у пациентов, получающих и не получающих ферментозаместительную терапию (ФЗТ), в исследовании [16] и в модели.
Рисунок 1. Вероятность отсутствия комбинированного исхода в зависимости от давности наблюдения у пациентов, получающих и не получающих ферментозаместительную терапию (ФЗТ), в исследовании [16] и в модели.
Figure 1. Probability of the combined outcome non-occurrence in patients receiving and not receiving enzyme replacement therapy (ERT) over the observation period; Data from [16] and from the proposed model.
была значительно ниже – всего 48%. Поскольку известно, что про- гноз БФ для женщин более благоприятен, чем для мужчин, в на- шей модели для сравнения были использованы данные по выжи- ваемости только для мужчин из когорты FOS. Так как в публикации была приведена кривая выживаемости для всей когорты FOS, наша модель была построена на приведенной частоте комбиниро- ванного исхода у мужчин (26% после 2-летнего периода терапии)
и предположении, что вероятность данного исхода остается все время одинаковой и не зависит от продолжительности терапии (то есть было использовано экспоненциальное распределение).
Горизонт моделирования составил 10 лет для оценки числа не- благоприятных исходов у пациентов с ранним и поздним началом ФЗТ. Для оценки числа исходов у пациентов, получающих и не по- лучающих ФЗТ (агалсидазу альфа), был выбран горизонт 5 лет,
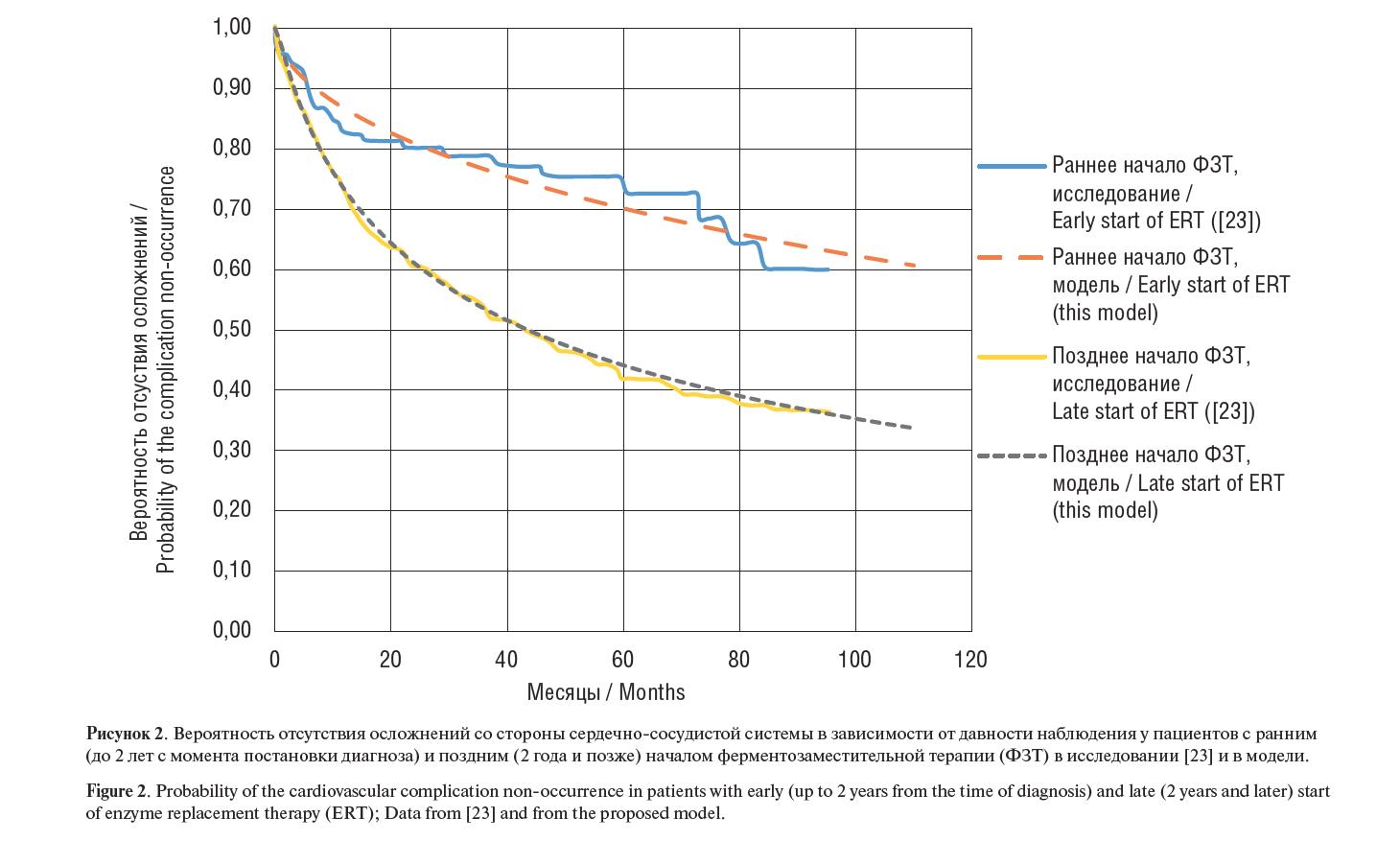
Рисунок 2. Вероятность отсутствия осложнений со стороны сердечно-сосудистой системы в зависимости от давности наблюдения у пациентов с ранним (до 2 лет с момента постановки диагноза) и поздним (2 года и позже) началом ферментозаместительной терапии (ФЗТ) в исследовании [23] и в модели.
Figure 2. Probability of the cardiovascular complication non-occurrence in patients with early (up to 2 years from the time of diagnosis) and late (2 years and later) start of enzyme replacement therapy (ERT); Data from [23] and from the proposed model.
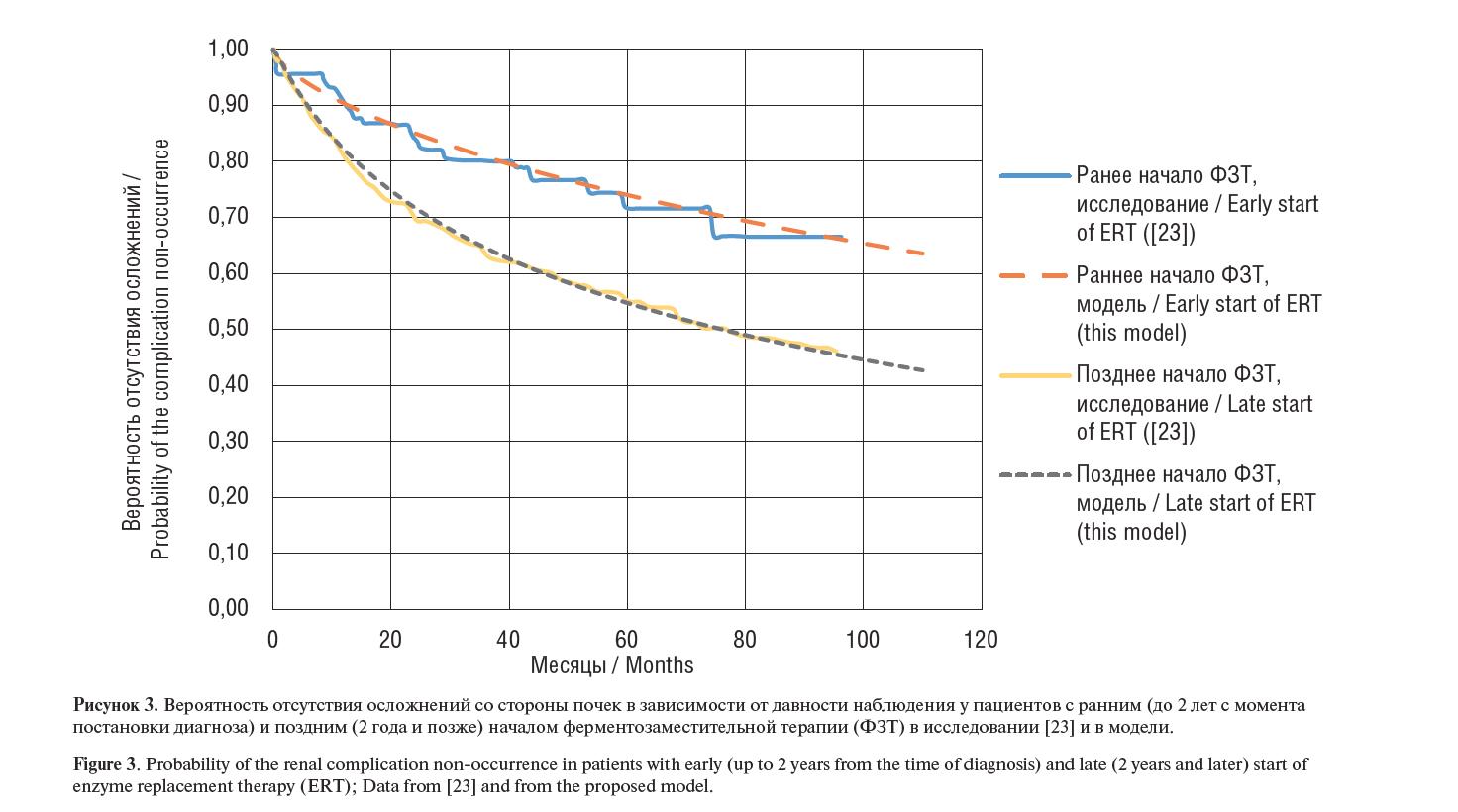 Рисунок 3. Вероятность отсутствия осложнений со стороны почек в зависимости от давности наблюдения у пациентов с ранним (до 2 лет с момента постановки диагноза) и поздним (2 года и позже) началом ферментозаместительной терапии (ФЗТ) в исследовании [23] и в модели.
Рисунок 3. Вероятность отсутствия осложнений со стороны почек в зависимости от давности наблюдения у пациентов с ранним (до 2 лет с момента постановки диагноза) и поздним (2 года и позже) началом ферментозаместительной терапии (ФЗТ) в исследовании [23] и в модели.
Figure 3. Probability of the renal complication non-occurrence in patients with early (up to 2 years from the time of diagnosis) and late (2 years and later) start of enzyme replacement therapy (ERT); Data from [23] and from the proposed model.
так как реальный период наблюдения, на основании которого строилась модель, был значительно короче, чем в исследовании, изучавшем исходы в зависимости от времени начала терапии.
Исходы моделировались для условной когорты в 100 человек. В построенных моделях для каждого года нахождения в модели определялось число пациентов, у которых развились изучаемые жизнеугрожающие исходы, и рассчитывалась разница между сравниваемыми группами.
Результаты
В результате моделирования были рассчитано число пациентов с БФ, у которых развились жизнеугрожающие осложнения в зави- симости от срока начала терапии агалсидазой альфа (табл. 2-3).
В таблице 4 представлены полученные нами результаты дополни- тельного моделирования, в котором сравнивались исходы для пациентов, получавших агалсидазу альфа и не получавших препа- раты ФЗТ.
Как видно из таблицы 2, раннее (в первые два года после появ- ления симптомов) назначение агалсидазы альфа позволяет до- полнительно предотвратить неблагоприятные сердечно-сосудис- тые события у 26,9% пациентов в течение10 лет проведения ФЗТ. Также раннее начало ФЗТ позволяет дополнительно предотвра- тить неблагоприятные почечные события у 20,8% пациентов в те- чение 10 лет терапии.
Следует отметить, что по результатам моделирования частота не- благоприятных событий со стороны сердечно-сосудистой системы
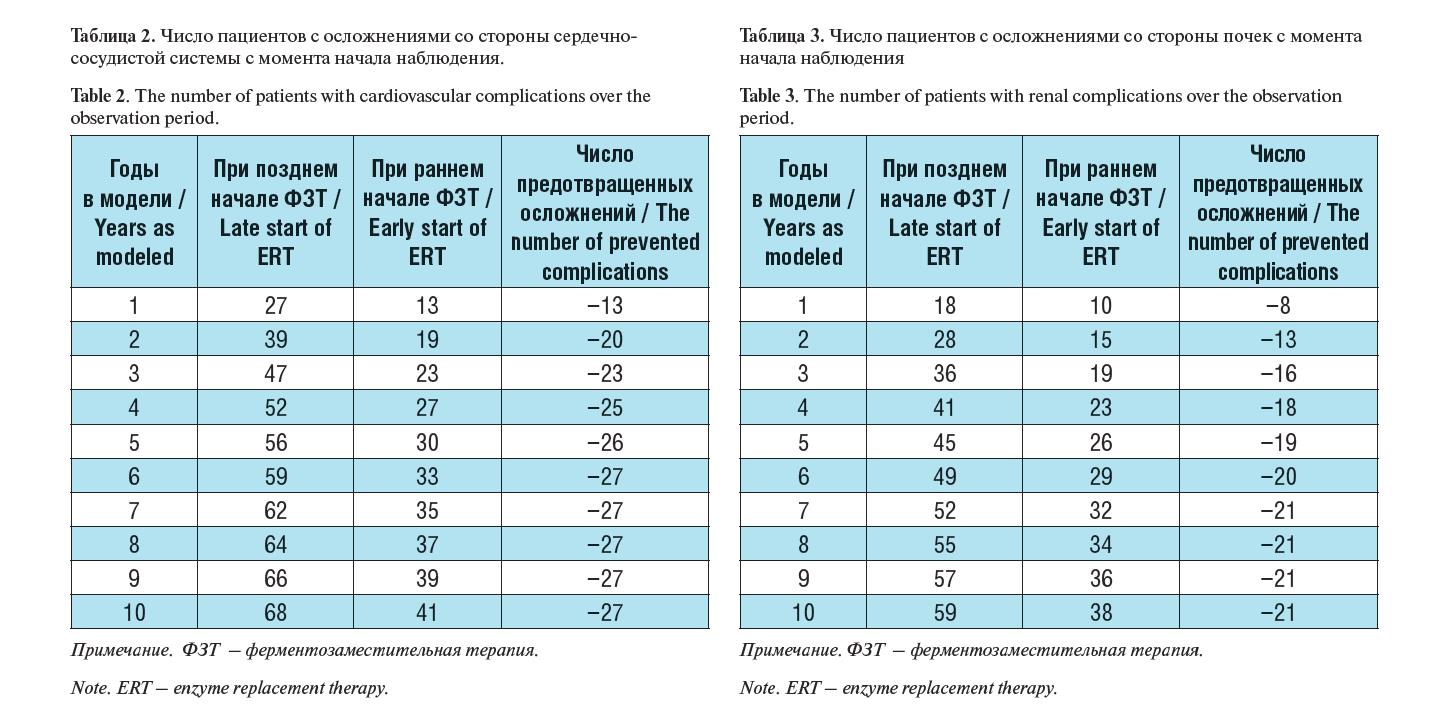
Таблица 2. Число пациентов с осложнениями со стороны сердечно- сосудистой системы с момента начала наблюдения.
Table 2. The number of patients with cardiovascular complications over the observation period.
Примечание. ФЗТ – ферментозаместительная терапия. Note. ERT – enzyme replacement therapy.
Таблица 3. Число пациентов с осложнениями со стороны почек с момента начала наблюдения
Table 3. The number of patients with renal complications over the observation period.
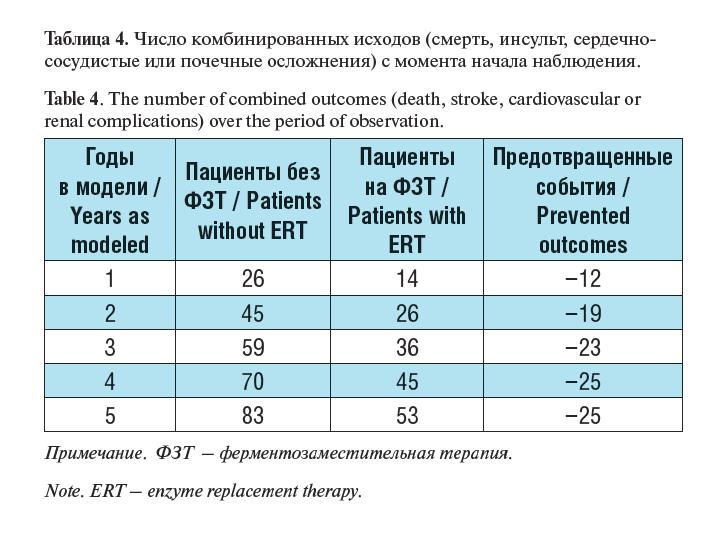 Таблица 4. Число комбинированных исходов (смерть, инсульт, сердечно- сосудистые или почечные осложнения) с момента начала наблюдения.
Таблица 4. Число комбинированных исходов (смерть, инсульт, сердечно- сосудистые или почечные осложнения) с момента начала наблюдения.
Table 4. The number of combined outcomes (death, stroke, cardiovascular or renal complications) over the period of observation.
и почек различается в группах пациентов приблизительно в 2 раза, при этом данная тенденция более выражена в первые 5 лет.
Результаты моделирования, представленные в таблице 4, по- зволяют сделать вывод о том, что назначение агалсидазы альфа в течение 5 лет позволяет предотвратить инсульт, сердечно-сосу- дистые, почечные осложнения, или смерть у 25% пациентов.
Обсуждение
В результате моделирования было продемонстрировано, что проведение ФЗТ в течение 5 лет может сократить число жизнеу- грожающих осложнений у пациентов с БФ на 25%. Также было показано, что раннее назначение препаратов ФЗТ (в течение двух лет после появления симптомов) позволяет значительно улучшить исходы пациентов с БФ и дополнительно (по сравнению с более поздним началом терапии) предотвратить основные осложнения более, чем у 20% пациентов. Существуют основания полагать, что помимо снижения частоты неблагоприятных событий, также бу- дет наблюдаться повышение качества жизни пациентов, в т.ч. за счет снижения болевых ощущений.
Следует отметить, что в нашем исследовании мы вынуждены были опираться на данных зарубежных регистров, а российская популяция может значительно отличаться из-за гораздо большего числа пациентов с уже развившимися осложнениями, в т.ч. из-за поздней постановки диагноза.
В настоящее время существуют очевидные сложности, связан- ные с несовершенством процесса выявления пациентов – прояв- ления БФ при отсутствии осведомленности о заболевании легко принять за симптомы иных заболеваний. Таким образом можно предполагать, что многие пациенты с БФ в РФ не имеют установ- ленного диагноза и не получают адекватного лечения. Для совер- шенствования процесса диагностики и увеличения доли пациен-
тов, заболевание у которых выявляется на ранних стадиях, необходимо повышение осведомленности врачей о БФ.
Проблема ведения пациентов с БФ в России связана с целым рядом причин, среди которых – низкая осведомленность врачей первичного звена о болезни Фабри, связанная с этим поздняя ди- агностика, отсутствие единой маршрутизации на региональном и федеральном уровне. Рекомендованная в федеральных центрах терапия БФ часто оспаривается либо не выполняется в регионах, что может быть связано с недостатком понимания проблемы БФ и дефицитом региональных бюджетов.
Пример с рядом других редких, хронических дорогостоящих за- болеваний, включенных в 2008 г. в программу «7 нозологий», рас- ширенную в 2018 г. еще на пять заболеваний, подтверждает, что доступ к дорогостоящей, но эффективной патогенетической тера- пии можно улучшить, перенеся финансирование лекарственного обеспечения на федеральный уровень.
В настоящем исследовании не проводились ни оценка затрат на проведение ФЗТ, ни сравнение затрат на ФЗТ с использовани- ем одного или другого препарата в силу того, что затраты на тера- пию во многом определяются массой тела пациента. Так, у паци- ентов с массой тела от 52,5 до 70 кг затраты за год терапии агалсидазой альфа составят 11,12 млн руб.2, а агалсидазой бета – 9,62 млн руб. В то же время у пациента с массой тела более 70, но менее 87,5 кг менее затратным является использование агалси- дазы альфа: год терапии будет стоить 13,91 млн руб. по сравне- нию с 14,43 млн руб. при назначении агалсидазы бета. Таким об- разом, при отсутствии доказательных данных о клиническом преимуществе одного или другого препарата лечащий врач дол- жен индивидуально выбирать препарат для пациента.
Проведение ФЗТ, как и любого другого патогенетического лече- ния при орфанных заболеваниях, требует значительных финансо- вых затрат, которые в настоящий момент возложены на ограни- ченные региональные бюджеты. Как следствие, для многих пациентов с БФ ограничивается доступ к ФЗТ, что приводит к тому, что терапия если и начинается, то со значительным опоз- данием, когда ее ценность для пациента снижается. При этом сле- дует отметить, что поскольку большинство пациентов с БФ явля- ются лицами трудоспособного возраста, в т.ч. в возрасте до 40 лет, задачу обеспечения доступа пациентов к лечению можно от- нести к стратегическим задачам системы здравоохранения РФ.
Заключение
Раннее начало ферментнозаместительной терапии у пациентов с болезнью Фабри позволяет существенно замедлить развитие у них тяжелых, жизнеугрожающих осложнений, увеличить про- должительность и повысить качество их жизни.
2 Все расчеты основаны на зарегистрированных инструкциях по ме- дицинскому применению препаратов и ценах в перечне жизненно необходимых и важнейших лекарственных препаратов, без учета торговых надбавок и НДС.
Литература:
- Brady R. O., Gal A. E., Bradley R. M., Martensson , Warshaw A. L., Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967; 276 (21): 1163-7.
- Kint J. A. Fabry’s disease: alpha-galactosidase deficiency. Science. 1970; 167 (3922): 1268-9.
- Мухин Н., Моисеев В., Моисеев С., Фомин В., Кобалава Ж., Пулин А. Диагностика и лечение болезни Фабри. Клиническая фармакология и терапия. 2013; 22 (2): 11-20.
- Кузенкова Л. М., Намазова-Баранова Л., Подклетнова Т., Ге- воркян А., Вашакмадзе Н., Савостьянов К., Студеникин В., Пуш- ков С. Болезнь Фабри: особенности заболевания у детей и под- ростков. Вопросы современной педиатрии. 2015; 14 (3): 341-8.
- Warnock G., West M. L. Diagnosis and management of kidney involvement in Fabry disease. Adv Chronic Kidney Dis. 2006; 13 (2): 138-47.
- Basic-Jukic , Kes P., Coric M., Basic-Kes V. Renal complications of Fabry disease. Curr Pharm Des. 2013; 19 (33): 6046-50.
- Desnick , Ioannou Y., Eng C. a-galactosidase A deficiency: Fabry disease. The metabolic and molecular bases of inherited disease. (eds Scriver CR, Beaudet AL, Sly WS, Valle D.) p3733-3774. Book
a-galactosidase A deficiency: Fabry disease. The metabolic and molecular bases of inherited disease. (eds Scriver CR, Beaudet AL, Sly WS, Valle D.) p3733-3774. Editor McGraw-Hill. New York. 2001.
- Beck Demographics of FOS – the Fabry Outcome Survey. Fabry Disease: Perspectives from 5 Years of FOS. Mehta A. et al. Oxford. 2006.
- Ginsberg Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey. Fabry Disease: Perspectives from 5 Years of FOS. Mehta A. et al. Oxford. 2006.
- Schiffmann R., Warnock D. G., Banikazemi M., Bultas , Linthorst G. E., Packman S., Sorensen S. A., Wilcox W. R., Desnick R. J. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24 (7): 2102-11.
- Федеральные клинические рекомендации по диагностике и лечению болезни Фабри. 2015 [Электронный ресурс] URL: http:// мороздгкб.рф/wp-content/uploads/2017/03/Федеральные-клини- ческие-рекомендации-по-диагностике-и-лечению-болезни-Фа- бри-2015г.pdf. Дата обращения: 06.2018.
- Mehta A., Beck M., Eyskens F., Feliciani C., Kantola I., Ramaswami , Rolfs A., Rivera A., Waldek S., Germain D. P. Fabry disease: a review of current management strategies. Qjm. 2010; 103 (9): 641-59.
- El Dib , Gomaa H., Carvalho R. P., Camargo S. E., Bazan R., Barretti P., Barreto F. C. Enzyme replacement therapy for Anderson- Fabry disease. Cochrane Database Syst Rev. 2016; 7: Cd006663.
- Schiffmann R., Kopp J. B., Austin H. A., 3rd, Sabnis , Moore D. F., Weibel T., Balow J. E., Brady R. O. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. Jama. 2001; 285 (21): 2743-9.
- Hughes A., Elliott P. M., Shah J., Zuckerman J., Coghlan G., Brookes J., Mehta A. B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double- blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008; 94 (2): 153-8.
- Banikazemi , Bultas J., Waldek S., Wilcox W. R., Whitley C. B., McDonald M., Finkel R., Packman S., Bichet D. G., Warnock D. G., Desnick R. J. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007; 146 (2): 77-86.
- Bierer , Balfe D., Wilcox W. R., Mosenifar Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J Inherit Metab Dis. 2006; 29 (4): 572-9.
- Eng M., Guffon N., Wilcox W. R., Germain D. P., Lee P., Waldek S., Caplan L., Linthorst G. E., Desnick R. J. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001; 345 (1 ): 9-16.
- Sirrs M., Bichet D. G., Casey R., Clarke J. T., Lemoine K., Doucette S., West M. L. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab. 2014; 111 (4): 499-506.
- Vedder C., Linthorst G. E., Houge G., Groener J. E., Ormel E. E., Bouma B. J., Aerts J. M., Hirth A., Hollak C. E. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. 2007; 2 (7): e598.
- Wyatt , Henley W., Anderson L., Anderson R., Nikolaou V., Stein K., Klinger L., Hughes D., Waldek S., Lachmann R., Mehta A., Vellodi A., Logan S. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol Assess. 2012; 16 (39): 1-543.
- Beck , Hughes D., Kampmann C., Larroque S., Mehta A., Pintos-Morell G., Ramaswami U., West M., Wijatyk A., Giugliani R. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis. Mol Genet Metab Rep. 2015; 3: 21-7.
- Linhart, A; Hughes, D; Gurevich, A; Joseph, A; Kerstens, R; Feriozzi, S; (2017) Prompt agalsidase alfa therapy initiation after symptom onset is associated with improved renal and cardiovascular outcomes in the Fabry Outcome Presented at: 13th Annual Research Meeting on We’re Organizing Research for Lysosomal Diseases (WORLD), San Diego, CA.
- Diaby , Adunlin G., Montero A. J. Survival modeling for the estimation of transition probabilities in model-based economic evaluations in the absence of individual patient data: a tutorial. Pharmacoeconomics. 2014; 32 (2): 101-8.
- Guyot , Ades A. E., Ouwens M. J., Welton N. J. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012; 12: 9.
References:
- Brady R. O., Gal A. E., Bradley R. M., Martensson , Warshaw A. L., Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967; 276 (21): 1163-7.
- Kint J. A. Fabry’s disease: alpha-galactosidase deficiency. Science. 1970; 167 (3922): 1268-9.
- Muhin , Moiseev V., Moiseev S., Fomin V., Kobalava Zh., Pulin A. Diagnosis and treatment of Fabry disease. klinicheskaja farmakologija i terapija (in Russian). 2013; 22 (2): 11-20.
- Kuzenkova L. M., Namazova-Baranova L., Podkletnova , Gevorkjan A., Vashakmadze N., Savost’janov K., Studenikin V., Pushkov S. Fabry disease: features of the disease in children and adolescents. Voprosy sovremennoj pediatrii (in Russian). 2015; 14 (3): 341-8.
- Warnock G., West M. L. Diagnosis and management of kidney involvement in Fabry disease. Adv Chronic kidney Dis. 2006; 13 (2): 138-47.
- Basic-Jukic N., Kes P., Coric M., Basic-Kes V. Renal complications of Fabry Curr Pharm Des. 2013; 19 (33): 6046- 50.
- Desnick , Ioannou Y., Eng C. a-galactosidase A deficiency: Fabry disease. The metabolic and molecular bases of inherited disease. (eds Scriver CR, Beaudet AL, Sly WS, Valle D.) p3733-3774. Book
a-galactosidase A deficiency: Fabry disease. The metabolic and molecular bases of inherited disease. (eds Scriver CR, Beaudet AL, Sly WS, Valle D.) p3733-3774. Editor McGraw-Hill. New York. 2001.
- Beck Demographics of FOS – the Fabry Outcome Survey. Fabry Disease: Perspectives from 5 Years of FOS. Mehta A. et al. Oxford. 2006.
- Ginsberg Nervous system manifestations of Fabry disease: data from FOS – the Fabry Outcome Survey. Fabry Disease: Perspectives from 5 Years of FOS. Mehta A. et al. Oxford. 2006.
- Schiffmann R., Warnock D. G., Banikazemi M., Bultas , Linthorst G. E., Packman S., Sorensen S. A., Wilcox W. R., Desnick R. J. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24 (7): 2102-11.
- Federal clinical guidelines for the diagnosis and treatment of Fabry disease. 2015 (in Russian) [Electronic resource] URL: http:// rf/wp-content/uploads/2017/03/Federal’nye-klinicheskie- rekomendacii-po-diagnostike-i-lecheniju-bolezni-Fabri-2015g.pdf. Accessed: 06.06.2018.
- Mehta A., Beck M., Eyskens F., Feliciani C., Kantola , Ramaswami U., Rolfs A., Rivera A., Waldek S., Germain D. P. Fabry disease: a review of current management strategies. Qjm. 2010; 103 (9): 641-59.
- El Dib , Gomaa H., Carvalho R. P., Camargo S. E., Bazan R., Barretti P., Barreto F. C. Enzyme replacement therapy for Anderson- Fabry disease. Cochrane Database Syst Rev. 2016; 7: Cd006663.
- Schiffmann R., Kopp J. B., Austin H. A., 3rd, Sabnis , Moore D. F., Weibel T., Balow J. E., Brady R. O. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. Jama. 2001; 285 (21): 2743-9.
- Hughes A., Elliott P. M., Shah J., Zuckerman J., Coghlan G., Brookes J., Mehta A. B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double- blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008; 94 (2): 153-8.
- Banikazemi , Bultas J., Waldek S., Wilcox W. R., Whitley C. B., McDonald M., Finkel R., Packman S., Bichet D. G., Warnock D. G., Desnick R. J. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007; 146 (2): 77-86.
- Bierer , Balfe D., Wilcox W. R., Mosenifar Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J Inherit Metab Dis. 2006; 29 (4): 572-9.
- Eng M., Guffon N., Wilcox W. R., Germain D. P., Lee P., Waldek S., Caplan L., Linthorst G. E., Desnick R. J. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001; 345 (1): 9-16.
- Sirrs M., Bichet D. G., Casey R., Clarke J. T., Lemoine K., Doucette S., West M. L. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab. 2014; 111 (4): 499-506.
- Vedder C., Linthorst G. E., Houge G., Groener J. E., Ormel E. E., Bouma B. J., Aerts J. M., Hirth A., Hollak C. E. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS one. 2007; 2 (7): e598.
- Wyatt , Henley W., Anderson L., Anderson R., Nikolaou V., Stein K., Klinger L., Hughes D., Waldek S., Lachmann R., Mehta A.,
Vellodi A., Logan S. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol Assess. 2012; 16 (39): 1-543.
- Beck M., Hughes D., Kampmann C., Larroque S., Mehta A.,
Pintos-Morell G., Ramaswami U., West M., Wijatyk A., Giugliani R. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis. Mol Genet Metab Rep. 2015; 3: 21-7.
- Linhart, A; Hughes, D; Gurevich, A; Joseph, A; Kerstens, R;
Feriozzi, S; (2017) Prompt agalsidase alfa therapy initiation after symptom onset is associated with improved renal and cardiovascular outcomes in the Fabry Outcome Survey. Presented at: 13th Annual Research Meeting on We’re Organizing Research for Lysosomal Diseases (WORLD), San Diego, CA.
- Diaby , Adunlin G., Montero A. J. Survival modeling for the estimation of transition probabilities in model-based economic evaluations in the absence of individual patient data: a tutorial. Pharmacoeconomics. 2014; 32 (2): 101-8.
- Guyot , Ades A. E., Ouwens M. J., Welton N. J. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012; 12: 9.
Сведения об авторах:
Игнатьева Виктория Игоревна – научный сотрудник Центра оценки технологий в здравоохранении Российской академии народного хозяйства и государственной службы при Президенте РФ. E-mail: ignateva@hta-rus.ru.
Моисеев Сергей Валентинович – д.м.н., заведующий кафедрой внутренних, профессиональных болезней и ревматологии ФГАОУ ВО «Первый МГМУ им. И. М. Сеченова» (Сеченовский университет).
Буланов Николай Михайлович – ассистент кафедры внутренних, профессиональных болезней и ревматологии ФГАОУ ВО «Первый МГМУ им. И. М. Сеченова» (Сеченовский университет).
Каровайкина Екатерина Александровна – ассистент кафедры внутренних, профессиональных болезней и ревматологии ФГАОУ ВО «Первый МГМУ им. И. М. Сеченова» (Сеченовский университет).
Моисеев Алексей Сергеевич – клинический ординатор факультета фундаментальной медицины МГУ им. М. В. Ломоносова.
About the authors:
Viktoria I. Ignatyeva – Researcher at the Center for Health Technology Assessment, Russian Presidential Academy of National Economy and Public Administra- tion. E-mail: ignateva@hta-rus.ru.
Sergey V. Moiseev – MD, Doctor of Medical Science, Head of the Department of Internal & Occupational Diseases and Rheumatology, Sechenov First Moscow State Medical University, Ministry of Healthcare of Russia.
Nikolay M. Bulanov – MD, Assistant, Department of Internal & Occupational Diseases and Rheumatology, Sechenov First Moscow State Medical University, Ministry of Healthcare of Russia
Ekaterina A. Karovaikina – MD, Assistant, Department of Internal & Occupational Diseases and Rheumatology, Sechenov First Moscow State Medical Universi- ty, Ministry of Healthcare of Russia.
Alexey S. Moiseev – Clinical Intern, Faculty of Fundamental Medicine, Lomonosov Moscow State University.





