
 Нарушения обмена цикла мочевины — это врожденные ошибки метаболизма, при которых, из-за частичной или полной инактивации соответствующих ферментов, нарушается образование малотоксичной мочевины, что приводит к острой или хронической интоксикации аммиаком — гипераммониемии.
Нарушения обмена цикла мочевины — это врожденные ошибки метаболизма, при которых, из-за частичной или полной инактивации соответствующих ферментов, нарушается образование малотоксичной мочевины, что приводит к острой или хронической интоксикации аммиаком — гипераммониемии.
Гипераммониемия (ГА) — клиническая ситуация с повышением уровня аммиака (NH3) и (аммония, NH4+) в крови выше 100 мкМоль/л у новорожденых и выше 50 мкМоль/л у более старших детей и взрослых.
ГА возникает вследствие нарушения детоксикации аммония в паренхиме печени и гиперпродукции – вследствие дефектов ферментов орнитинового цикла (цикла образования мочевины, ЦОМ). При генетически детерминиованной энзиматической недостаточности одного из ферментов ЦОМ формируется первичная гипераммониемия, при печеночной недостаточности как острой, хронической, так и декомпенсации цирроза возникает вторичная гипераммониемия с накоплением других токсичных метаболитов, кроме аммония, что клинически проявляется специфическим сладковато-гнилостным печеночным запахом.
Главным источником аммиака являются аминокислоты, белки, NH4+ представляет собой бесцветный газ, катион, конечный продукт азотистого обмена у теплокровных.
В процессе онтогенеза и перехода из водной среды обитания повышение концентраций аммиака стало небезопасным и потребовались другие пути выведения азотистых шлаков (аммония и мочевой кислоты, преимущественно у рыб, рептилий и земноводных) из организма, а именно мочевины, креатина, креатинина. Мочевина — основной конечный продукт азотистого обмена, в составе которого из организма выделяется до 90% всего выводимого азота, экскреция мочевины в норме составляет ∼25 г/сут.
Так как аммоний с легкостью проникает через тканевой барьер, и обычно концентрация в тканях в 10 раз превышает концентрацию в сыворотке крови, наиболее чувствительна к повреждению аммонием центральная нервная система, аммоний считается нейротоксином, действуя опосредованно через систему синтеза глютамина, накапливается и повреждает глиальные клетки, изменяет (повышает) осмолярность и осложняется отеком мозга. Патогенный эффект ГА зависит от продолжительности и тяжести интоксикации.
Представление об обмене аммиака, мочевины и других белковых метаболитах малоизвестно врачам по причине отсутствия доступной диагностики, лечения. Но по существу гипераммониемические состояния могут встретиться в практике врачей разных специальностей – от неонатологов, педиатров, неврологов, врачей скорой помощи, реаниматологов, гастроэнтерологов, гепатологов, токсикологов и др. Определение уровня аммония не входит в повседневную практику, поиск лаборатории, где можно определить аммоний, даже в областных городах представляет значительные трудности.
После приёма пищи из кишечника в плазму крови поступает много аминокислот, причём преобладают аминокислоты с разветвлённой боковой цепью (до 20% от общего количества), которые затем поглощаются, в основном, печенью, мышцами и мозгом. В мышцах происходит усиленный катаболизм этих аминокислот, причём они выступают основными донорами аминогруппы в синтезе аланина из пирувата .
В постабсорбтивном периоде основными источниками свободных аминокислот являются мышцы. Они поставляют в основном аланин и глутамин . Аланин поглощается печенью, глутамин — кишечником и почками. В кишечнике азот глутамина переносится в аланин или серин и в их составе транспортируется в печень, где активируется процесс глюконеогенеза. Таким образом, аланин и серин — основные гликогенные аминокислоты. Интенсивность глюконеогенеза из этих аминокислот намного выше, чем из всех других. На ограничении поступления с пищей аминокислот с разветвленной цепью основывается низкобелковая диете при БЦОМ.
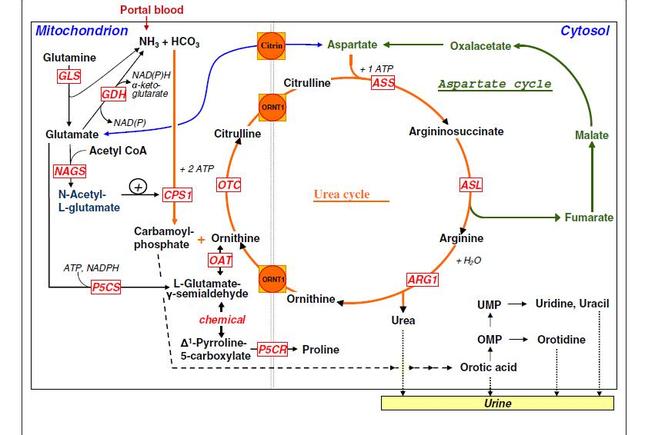
Рис.1. Болезни орнитинового цикла и место соответствующих ферментных дефектов (нестандартные аббревиатуры GDH глютаматдегидрогеназа, NAD(P) никотинамид аденин динуклеотид (фосфат), OAT орнитин аминотрансфераза, ОМP оротидин монофосфат, PSCR пирролин-5-карбоксилат редуктаза, PSCS- Δ1-пирролин -5-карбоксилат синтетаза, UMP- уридин монофосфат).
Основной реакцией обезвреживания аммиака в тканях является синтез глутамина, который затем используется в анаболических процессах и для обезвреживания веществ в печени. Ферменты глутаматдегидрогеназа и глутаминсинтетаза являются регуляторными и обусловливают скорость процессов образования и обезвреживания аммиака.
Таблица 1. Номенклатура болезней цикла образования мочевины
| Название | Аббревиатура | Ген | Локализация мутации | Тип наследования | OMIM | |
| 1 | N ацетил глютамат синтетазная недостаточность | NAGS дефицит | NAGS | 17q21.31 | AR | 237210
608300 |
| 2 | Карбомоилфосфат-синтетазная недостаточность | СPS1 дефицит | CPS1 | 2q35 | AR | 237300
608307 |
| 3 | Орнитинтранскарбамилазная недостаточность | OTC
дефицит |
OTC | Xp11.4 | X- сцепленное | 311250
300461 |
| 4 | Цитруллинемия тип1 | CTLN1, Аргинин-сукцинат
синтетазная недостаточность |
ASS1 | 9q34.11 | AR | 215700
603470 |
| 5 | Аргининянтарная ацидурия | Аргинин сукцинат лиазная недостаточность | ASL дефицит | 7q11.21 | AR | 207900
608310 |
| 6 | аргининемия | Дефицит аргиназы 1 | ARG1 дефицит | 6q23.2 | AR | 207800
608313 |
| 7 | HHH синдром
(гипераммониемия, гиперорнитинемия, гомоцитруллинурия) |
ORNT1 дефицит (митохондриальный дефицит орнитина, SLC25A15 | ORNT1 | 13q14.11 | AR | 238970 |
| 8 | Цитруллинемия тип 2 | Недостаточность цитрина, переносчик аспартат-глютамат | СTLN2 | 7q21.3 SLC25A13 | AR | 605814
603471 |
| 9 | Недостаточность глютамин синтетазы | GS недостаточность | GLUL | 1q25.3 | AR | 610015 |
| 10 | Транзиторная гипераммониемия новорожденных | THAN | AR | Ранняя инфантильная форма | ||
| 11 | Пирролин-5-карбоксилат синтетазная недостаточность | P5CS недостаточность | ALDH18A1 | 10q24.1 | AR | 219150 |
Различают две группы первичных гипераммониемий в зависимости от места энзиматического дефекта – прокисмальную до включения в орнитиновый цикл (в митохондриях – CPS1, NAGS и OTC) и дистальную – непосредственно протекающие в цитоплазме гепатоцитов в ЦОМ, (ASS, ASL, ARG, HHH) и два трансмембранных транспортера орнитина и цитруллина, вызывающих ННН синдром и недостаточность цитрина (ORNT1, CIT 2). См. рис 1
В РФ частота не установлена, тем более учитывая вариабельность течения болезней ЦОМ с дебютом в разные возрастные периоды от периода новорожденности (наиболее тяжелые, часто фатальные формы), младенчества, обычно связанные с введением белкового прикорма, у взрослых, обычно маскируются/проявляются неврологическими и психиатрическими симптомами.
Подавляющее большинство БЦОМ характеризуются аутосомнорецессивным типом наследования, за исключением ОТС – с Х-сцепленным типом наследования.
Кроме ферментного дефицита существует еще две болезни, обусловленные нарушением переноса субстратов через митохондриальную мембрану — дефицит транспортера орнитина 1 (орнитинтранслоказа -ORNT1) и дефицит цитрина (транспортера глютамата-аспартата).
Общая распространенность болезней ЦОМ, 1:8 000. Из них: ОТС 1:14 000, ASS 1:57 000, СPS 1:62 000, ASL 1:70 000, Дефицит аргиназы 1: 363 000. По данным Summar ML (2013), проанализировавшем результаты скрининга более 6 миллионов новорожденных некоторых штатов в США за 2001-2012 гг. распространенность БЦОМ получилась следующая: NAGSдефицит <1 :2000000, CPS1 дефицит 1:1300000, OTCдефицит 1:56 500, ASS1 дефицит 1:250000, ASL дефицит 1:218750, ARG дефицит 1:950000.
Недостаточность CPS1, ASS1, ASL, NAGS, и ARG наследуются по аутосомно-рецессивному образу. CPS1 дефицит и NAGS недостаточность являются наиболее тяжелыми из расстройств цикла мочевины. Лица, с полной недостаточностью CPS1 быстро развивают гипераммониемию в период новорожденности.
OTC дефицит типичен для мальчиков. Примерно 15% женщин-носителей мутации с лайонизацией второй Х-хромосомы развивают гипераммониемию в течение жизни, и многие из них требуют лечения.
Клинические характеристики
Принципиальные отличия клинической симптоматики определяются не только генетическим дефектом, активностью фермента, но в большей степени уровнем аммония и незрелостью ЦНС. Неонатальный и младенческий дебют протекает значимо тяжелее, чем у детей старше 3 лет и взрослых.
Для всех форм ГА типично нестабильность терморегуляции, склонность к гипотермии у новорожденных, у старших детей этот симптом не выявляется, типичны для всех форм кома и задержка в развитии, острые энцефалопатические кризы, судороги, атаксия, инсультоподобные эпизоды, рвота, тошнота, реакция на белковую пищу (у многих детей формируются пищевые предпочтения с отказом от белковой пищи).
Таблица 2. Особенности клинических проявлений при остром и хроническом течении БЦОМ
| Острое течение (метаболический криз) | Хроническое течение |
| От летаргии, сомноленции до комы, по типу энцефалита или лекарственной интоксикации
Острая энцефалопатия Судороги, Атаксия Инсультоподобные эпизоды Проходящая потеря зрения Рвота и прогрессирующая потеря аппетита Печеночная недостаточность Полиорганная недостаточность Циркуляторная периферическая недостаточность Пост-партум психоз Психоз, галлюцинации, паранойя, мании, эмоциональные и личностные изменения Новорожденные: Сепсис-подобная картина, температурная нестабильность, гипервентиляция, респираторный дистресс-синдром |
Спутанность сознания, летаргия, головокружение
Головная боль, мигренеподобные атаки, тремор, атаксия, дизартрия Тахипное Астериксис (хлопающий тремор) у взрослых Трудности/невозможность обучения, задержка моторного и интеллектуального развития умственная отсталость Хорея, церебральный паралич Корковая потеря зрения Отвращение к белку, самостоятельный выбор диеты с низким содержанием белка |
- выделены симптомы, встречающиеся у подавляющего большинства больных





