
 Расширение наших знаний в области медицины, позволяет понимать механизм развития того или иного заболевания у человека. Например, лизосомные болезни, связанные с нарушением обмена мукополисахоридов. Впервые попытка классифицировать эту группу заболеваний до открытия биохимических основ их развития была сделана английским врачом Эллисом который ввел в клинику термин «гаргоилизм» в 1936 году, который объединил мукополисахаридозы типа I (Н, S, H/S) и типа II (синдром Хантера). В последующем, по мере накопления знаний о течении этих заболеваний и расширения лабораторных возможностей, понятие о данном заболевании расширилось до 6 типов, а каждый тип имеет несколько подтипов [1, 2]. Причем каждый подтип данного заболевания имеет изученный биохимический дефект и на 3 типа имеется ферментзаместительная терапия. Благодаря этому развитию в медицине английская фраза orphan disease дала название группе заболеваний, которые считаются редкими. Проблема редких — «орфанных болезней» стоит перед здравоохранением многих стран мира потому, что медицина находится в переходном этапе от привычной нам медицины к новому этапу высокотехнологичной персонифицированной медицины.
Расширение наших знаний в области медицины, позволяет понимать механизм развития того или иного заболевания у человека. Например, лизосомные болезни, связанные с нарушением обмена мукополисахоридов. Впервые попытка классифицировать эту группу заболеваний до открытия биохимических основ их развития была сделана английским врачом Эллисом который ввел в клинику термин «гаргоилизм» в 1936 году, который объединил мукополисахаридозы типа I (Н, S, H/S) и типа II (синдром Хантера). В последующем, по мере накопления знаний о течении этих заболеваний и расширения лабораторных возможностей, понятие о данном заболевании расширилось до 6 типов, а каждый тип имеет несколько подтипов [1, 2]. Причем каждый подтип данного заболевания имеет изученный биохимический дефект и на 3 типа имеется ферментзаместительная терапия. Благодаря этому развитию в медицине английская фраза orphan disease дала название группе заболеваний, которые считаются редкими. Проблема редких — «орфанных болезней» стоит перед здравоохранением многих стран мира потому, что медицина находится в переходном этапе от привычной нам медицины к новому этапу высокотехнологичной персонифицированной медицины.
На сегодняшний день складывается ложное впечатление о редкости данных заболеваний ведь каждое в отдельности встречается в популяции не чаще чем 1 на 10 тысяч человек. Каждая в отдельности нозологическая форма заболевания может иметь низкую распространенность, однако общее число больных орфанными заболеваниями, а их на данный момент регламентировано 24 нозологические формы, даже при приблизительном подсчете их оказывается достаточно велико 1 на 500 и это если не брать во внимание, что данную группу планируется увеличить.
С развитием науки меняются и образовательные стандарты для обучения новых медицинских кадров, вводятся новые подходы к ведению пациентов. В этих быстро изменяющихся условиях требуется регулярная переработка подходов к ведению данных пациентов, их маршрутизации, алгоритму обследования, постановки диагноза и назначения комплексного индивидуального лечения. Сегодняшняя реальность заключается в том, что специалистам, с большим стажем работы трудно приспосабливаться к изменяющимся условиям. Поэтому в большинстве случаев редкие заболевания не удается диагностировать вовремя. Этому способствуют плохая осведомленность врачей о клиническом течении редких заболеваний, отсутствие четких рекомендаций врачу, заподозрившему редкое заболевание и отсутствие алгоритма ведения такого пациента. Можно выделить организационные и медицинские проблемы редких болезней:Трудности диагностики; отсутствие опыта и знаний у врачей; Проблемы лечения и реабилитации больных; Стандарты лечения редких болезней; Определения группы высокого риска по ожиданию больного орфанным заболеванием; Проблемы носителей патологического гена, их обследование, тактика ведения, предгравидарная подготовка и, возможно, вспомогательные репродуктивные технологии; Выстраивание преемственности между специалистами в оказании комплексной помощи данным пациентам; Проблемы клинических испытаний лекарств для больных; механизм закупок лекарственных средств для орфанных болезней; Механизмы включения новых больных, выявленных в процессе диагностики; Прогнозирование числа ожидаемых больных; Преемственность между специалистами при переходе больных с редкими болезнями от педиатров к терапевтам; Образовательные программы по редким болезням; Психологические, семейные, социальные, финансовые.
Редкие болезни условно можно разделить на две группы: немоногенные и моногенные. К немоногенным орфанным заболеваниям можно отнести: гемолитико-уремический синдром, пароксизмальная ночная гемоглобинурия (Маркиафавы-Микели), апластическая анемия неуточненная, идиопатическая тромбоцитопеническая пурпура (синдром Эванса), преждевременная половая зрелость центрального происхождения, легочная (артериальная) гипертензия (идиопатическая) (первичная), юношеский артрит с системным началом. К моногенным орфанным заболеваниям можно отнести: гемолитико-уремический синдром (наследственный), наследственный дефицит факторов II (фибриногена), VII (лабильного), X (Стюарта-Прауэра), дефект в системе комплемента, нарушения обмена ароматических аминокислот (классическая фенилкетонурия, другие виды гиперфенилаланинемии), тирозинемия, болезнь «кленового сиропа», другие виды нарушений обмена аминокислот с разветвленной цепью (изовалериановая ацидемия, метилмалоновая ацидемия, пропионовая ацидемия), нарушения обмена жирных кислот, гомоцистинурия, глютарикацидурия, галактоземия, другие сфинголипидозы: болезнь Фабри (Фабри-Андерсона), Нимана-Пика, мукополисахаридозы I, II, VI типов, острая перемежающая (печеночная) порфирия, нарушения обмена меди (болезнь Вильсона), незавершенный остеогенез [4].
Данное разделение принципиально, так как в маршрутизацию пациента в обязательном порядке должна включаться вся семья выявленного пациента. На сегодняшний момент при введении по душевого финансирования должно быть административное понимание о риска появления в данной семье повторного пациента с этой патологией. Причем круг риска обязательно должен устанавливать врач-генетик. Приведу пример родословной семьи К. с 2 пациентами с орфанным заболеванием с Х-сцепленным реццесивным типом наследования.
Родословная семьи К. с 2 пациентами с орфанным заболеванием с Х-сцепленным рецессивным типом наследования (рис.1).

На учете в медико-генетическом отделе находился пациент из 5-го поколения, при детальном сборе семейного анамнеза был выявлен ещё 1 пациент из 6-го поколения.
Еще одной трудностью в постановке диагноза становится полиморфизм клинической картины заболевания. Полиморфизм часто зависит от экспрессии гена в том или ином генотипе. Иногда врачу клиницисту трудно собрать весь симптомокомплекс в одном пациенте, а генетик работая с семьей может собрать воедино все клинические проявления, основываясь на данных о нескольких членах семьи. Как правило, заболевания из группы моногенных, связаны с дефектом одного гена, кодирующего индивидуальный фермент, который обеспечивает превращение одного вещества (субстрат) в другое (продукт). В большинстве случаев таких расстройств патогенным является накопление веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений. По мере накопления патологических соединений в организме проявляется клиническая картина. Трудности в диагностики заключаются в том что, клинические симптомы, как правило, неспецифичны; многие заболевания чрезвычайно сходны по клиническим проявлениям; точная диагностика возможна только с помощью сложных современных лабораторных методов. Поэтому в алгоритм для врачей, особенно работающих с детьми, должен быть прописан круг симптомов, при появлении которых в дифференциальный диагноз должны включаться заболевания из группы орфанных.
Признаки, требующие исключения орфанного заболевания в неонатальном периоде и на первом году жизни:
1. Рвота, дегидротация, желтуха, мышечная гипо и гипертония, нарушение дыхания, судороги, летаргия, кома, асцит, необычный запах мочи (исключить ВПР ЖКТ).
2. Диарея, гипотрофия (исключить экзогенные причины).
3. Гепато, сплено, гепатоспленомегалия.
4. Проградиентно развившаяся к году жизни умственная отсталость, мышечная гипо и гипертония, судорожные припадки, развившиеся в первые месяцы жизни.
5. Лабораторные данные: метаболический ацидоз, алкалоз, гипогликемия, сахар, белок, ацетон в моче.
После первого года жизни: Умственная отсталость (УО) неясной этиологии; УО с задержкой физического развития, судорожными припадками, интоксикацией, летаргией, коматозными состоянием, рвотой, диареей, поражением печени, почек, экземой, вывихом, подвывихом хрусталика, глухотой, алалией, дизлексией; Сочетание УО с низким ростом, грубыми чертами лица, гидроцефальным черепом, тугоподвижностью суставов, сколиозом, гепатоспленомегалией, помутнением роговицы, тугоухостью; Проградиентное развитие УО и неврологической симптоматики после периода нормального развития разной длительности.
• Гипотрофия неясной этиологии.
• Непереносимость отдельных видов пищи, синдром мальабсорбции.
• Нефролитиаз у детей.
Это позволит вовремя выявить патологические нарушения и начать лечение. Очень важно понимать, что орфанное заболевание как правило с полисистемными проявлениями, поэтому важно комплексное обследование с привлечением междисциплинарной команды специалистов. Оптимального единого алгоритма не существует, так как у каждого пациента будет индивидуальное течение заболевания даже внутри одной нозологии. Поэтому при подозрении на орфанное заболевание врачу пройдется по основному симтомакомплексу определиться с наиболее вероятным заболеванием из этой группы, учитывая особенности патогенеза болезни, возраст и состояние ребенка и в первую очередь выполнить биохимический поиск на отсутствие или недостаток определенного фермента.
Только при обнаружении биохимического дефекта можно говорить о наличии орфанного заболевания и вот на этом этапе должен быть, обязательно подключен к междисциплинарной команде врач-генетик. Он определяется и с объемом дальнейших генетических исследований и кругом лиц в данной семье которым необходимы генетические обследования. Это позволит оказать помощь не только данному пациенту, но и найти других пациентов в этой семье с данным заболеванием на доклиническом этапе. Ведь своевременное адекватное лечение в этом случае позволит избежать осложнений, улучшит качество и увеличит продолжительность жизни пациентов. Для каждого пациента подбирается соответствующая медикаментозная терапия. При отсутствии или дефиците какого-либо фермента назначается ферментозаместительная терапия, которую необходимо начинать как можно раньше – до развития необратимых изменений.
Вопросы целесообразности и своевременности начала лечения это тоже серьёзная проблема как с морально-этической точки зрения, так и с экономической.
В ныне существующих регламентирующих документах в разделах лечение конкретно прописано заболевание и препарат/ы рекомендуемые для назначения пациенту при конкретной нозологической форме и рекомендуемый объем введения данного препарата. Эти рекомендации не предусматривают возможности отклонения от установленных правил на местах. Это часто вызывает вопросы у специалистов по профилю. Приведу пример клинического полиморфизма течения 2-х пациентов с болезнью Фабри.
Болезнь Фабри, клинический полиморфизм
| СИМПТОМЫ | 1-Й ПАЦИЕНТ | 2-Й ПАЦИЕНТ |
| Возраст начала | 8 лет | 16 лет |
| Первое проявление | Парастезии | Ангиокератомы |
| Патология ЖКТ | есть | есть |
| Патология почек | С 29 лет, ХПН 1 С 32 лет на гемодиализе |
нет |
| Состояние | Смерть в 36 лет | 35 лет, самочувствие удовлетворительное |
Опыт лечения пациентов в мировом медицинском сообществе неоднозначно утверждает единую дозировку введения препарата, начало введения препарата и возможность корректировки дозы. Нужно рассмотреть возможность на территории введения междисциплинарного консилиума для решения вопроса оценки тяжести пациента, эффективности проводимого лечения, и изменении дозы препарата в зависимости от изменяющейся клинической и лабораторной картины, как в лучшую, так и в худшую стороны. Этот консилиум должен иметь легитимность в лице Министерства Здравоохранения РФ, назначаться консилиум должен приказом по конкретной нозологической форме орфанного заболевания приказом местного органа здравоохранения, так как обеспечение лекарствами пациентов с редкими заболеваниями, согласно действующему законодательству осуществляется за счет средств регионального бюджета. Данный консилиум должен определять и очередность взятия пациента на лечение. Так как в режиме дефицита бюджета могут возникнуть трудности с быстрым предоставлением денежных средств на приобретение препарата. На сегодняшний момент процедуры очередности на выделения денежных средств не прописана ни в одном законодательном акте. Так же не решен вопрос о возможности снятия пациента с орфанным заболеванием с лечения. Не разработаны критерии оценки эффективности препарата, особенно если пациент является инвалидом с вовлечением центральной нервной системы лечение начато на поздней стадии развития заболевания и на протяжении нескольких лет получения дорогостоящего лечения его общее состояние не только не улучшается, но даже объективно ухудшается.
В медицинских кругах все больше задается вопрос о целесообразности введения неонатального скрининга на орфанные заболевания. Проводить генетическое тестирование всем новорожденным слишком дорого даже для обеспеченных систем здравоохранения. Хотя, безусловно, расширение программы неонатального скрининга позволило бы улучшить ситуацию по редким заболеваниям в стране. Но вопросы скрининга ведут за собой рад административных решений: покупка оборудования на территории для скрининга всех новорожденных. Это возможно только на тандемном масс-спектрометре [3]. На нем можно уже сегодня посмотреть более 30 заболеваний одномоментно. Но возникает следующий вопрос об обучении специалистов способных работать на данной аппаратуре и возможности правильной интерпретации полученных результатов. При наличии такой возможности на территории должна быть развита и ДНК диагностика (как подтверждающая диагностика), а это возможно только на ДНК-секвенаторе. Только приобретение такого комплекса оборудования и его запуск обойдутся региональному бюджету в сумму около 150 миллионов рублей. Можно пойти по пути заключения договоров с НИИ, имеющими данную аппаратуру на территории с покупкой реактивов на нужды здравоохранения региона, возможен и вариант заключения договора с Федеральным центром на оказание данных услуг для региона.
Еще одной проблемой являются носители патологичного гена, их обследование, тактика ведения, предгравидарная подготовка и, возможно, вспомогательные репродуктивные технологии.
Как пример можно привести семью Д. с Х-сцепленным рецессивным орфанным заболеванием.
Семья Д. с Х-сцепленным рецессивным орфанным заболеванием (рис. 2).
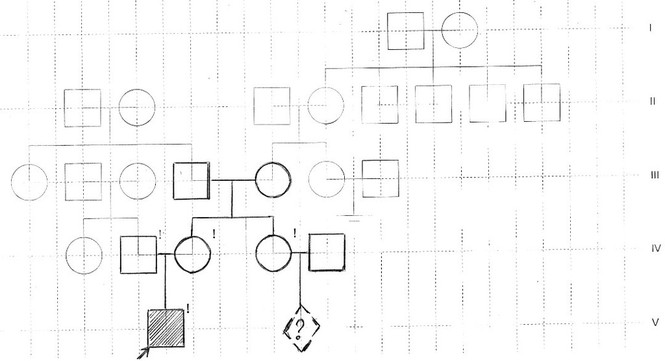
В маршрутизации этих пациентов должна быть статья расходов на ДНК-диагностику у кровных родственников из группы риска по рождению детей с орфанным заболеванием. Как вариант для больного с орфанным заболеванием и для его родственников (потенциальных носителей мутации) разработать механизм включения таких людей в региональные программы оказания помощи с помощью вспомогательных методов репродуктивной медицины с доимплантационной диагностикой по конкретной орфанной патологии.
На сегодня имеется еще несколько трудностей. Даже на этапе ведения регионального сегмента Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности. В приказе Министерства здравоохранения РФ от 19 ноября 2012г. за №950н «О формах документов для ведения регионального сегмента Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности» указано, что заполнение формы ведется врачом, к которому административно прикреплен пациент. В самом начале заполнения регистра это привело к путанице, что считать документом подтверждающим диагноз? Поэтому в регистр закрались ошибки: в частности, технические – врач перепутал диагноз и в регистр был занесен пациент с диагнозом, установленным в частном центре. При вызове данного пациента на консилиум и его дообследовании диагноз был снят. Поэтому на территории Новосибирской области было принято административное решение, что подача документов на включение в регистр производится только при наличии заключения главного специалиста по профилю орфанного заболевания.
Для прозрачной маршрутизации пациентов с орфанными заболеваниями утвердить необходимый общий перечень документов и административных действий, определить ответственных на каждом этапе за пациента с орфанным заболеванием от мысли об орфанном заболевании у пациента, до выработки тактики ведения и лечения данного пациента. Можно рассмотреть предполагаемую тактику в отношении к пациентам с орфанными заболеваниями:
1. Ввести перечень симптомов, при которых специалист обязан в круг дифференциальной диагностики включить орфанные заболевания (для моногенных орфанных заболеваний);
2. При подозрении или уверенности врача в наличии у пациента орфанного заболевания, врач обязан поставить об этом в известность главного профильного специалиста;
3. Главным определить профильного специалиста при системе здравоохранения в регионе по конкретной нозологической форме из группы орфанных заболеваний. Именно профильный специалист определяет дальнейшую тактику обследования или дообследования пациента и принятия решения о назначении лечения конкретного пациента. Данное решение должно приниматься профельным специалистом на основании заключения из стационара, а так же выписки из амбулаторной карты пациента с места жительства (заверенная главным врачом учреждения), из оценки общего состояния пациента, с учетом этиологии развития данного заболевания, или основываясь на заключении из федерального центра;
4. При установлении у пациента орфанного заболевания главный профильный специалист удостоверяет на своем бланке диагноз и назначения терапии с указанием медикоментозного препарата и его дозировки необходимой для лечения данного пациента;
5. В спорных ситуациях или в случае затруднения при установлении диагноза на территории главный профильный специалист может воспользоваться региональной квотой в учреждении федерального уровня для дообследования пациента и выработки тактики ведения данного пациента или созвать консилиум из смежных специалистов для решения вопроса о выработки дальнейшей тактики по конкретному пациенту.
6. На основании заключения главного профильного специалиста врач поликлинического звена, к которому прикреплен пациент заполняет форму установленную приказом МЗ от 19 ноября 2012г. за №950н «О формах документов для ведения регионального сегмента Федерального регистра лиц, страдающих жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, приводящими к сокращению продолжительности жизни граждан или их инвалидности» о включение данного пациента в региональный сегмент Федерального регистра;
7. Для четкой работы в поликлиническом звене с пациентами с установленным орфанным заболеванием рекомендовано маркировать титульный лист амбулаторной карты пациента (основной диагноз, номер в регистре, дата включения в регистр);
8. Для моногенных орфанных заболеваний должно быть 2 профильных главных специалиста: специалист по генетике и специалист по основному клиническому проявлению.
9. Для запуска процедуры выделения денежных средств для закупки препарата необходимого пациенту должен быть утвержден перечень документов, предоставляемый в отдел лекарственного обеспечения.
Список литературы:
1.Кеннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. Атлас-справочник. Пер. с англ. – Практика, 2011. 1024 с.; 2. Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. Атлас-справочник. 3-е издание. 2007. 448 с.; 3. Краснопольская К.Д. Наследственные болезни обмена веществ.; 4. www.omim.org





