
 Коллектив авторов: ФГБУ «Московский НИИ педиатрии и детской хирургии» Минздрава России (проф. П.В.Новиков, д.м.н. А.Н. Семячкина, д.м.н. В.Ю. Воинова), ФГБУ «Медико-генетический научный центр РАМН» (д.м.н. Е.Ю. Захарова, к.б.н. Е.Ю. Воскобоева).
Коллектив авторов: ФГБУ «Московский НИИ педиатрии и детской хирургии» Минздрава России (проф. П.В.Новиков, д.м.н. А.Н. Семячкина, д.м.н. В.Ю. Воинова), ФГБУ «Медико-генетический научный центр РАМН» (д.м.н. Е.Ю. Захарова, к.б.н. Е.Ю. Воскобоева).
Методология
Методы, использованные для сбора/селекции доказательств.
Доказательной базой для рекомендаций являются поиск в электронных базах данных PubMed и Mеdline. Данные клинические рекомендации подготовлены на основании международных клинических рекомендаций о диагностике и тактике ведения мукополисахаридоза, типа VI (MPSVI), разработанных группой экспертов по мукополисахаридозу VI типа, а также обзора публикаций клинических исследований. Глубина поиска составила 5 лет.
Методы, использованные для оценки качества и силы доказательств: оценка значимости в соответствии с рейтинговой схемой для оценки силы рекомендаций: мета-анализы высокого качества, систематизированные обзоры рандомизированных контролируемых исследований с низким риском систематических ошибок. Уровень доказательности 1++ -1+. Сила рекомендаций оценивалась по Оксфордской шкале — тип А. Для оценки качества, силы доказательств и формулирования рекомендаций использовался консенсус экспертов.
Индикаторы доброкачественной практики (Good Practice Points-GPPs) базировались на клиническом опыте рабочей группы по разработке рекомендаций.
Экономический анализ и публикации по фармакоэкономике не анализировались. Валидизация рекомендаций базировалась на внутренней экспертной оценке. Комментарии, полученные от экспертов, систематизировались и обсуждались председателем и членами рабочей группы. Проект рекомендаций был рецензирован независимыми экспертами.
Для окончательной редакции и контроля качества рекомендации были повторно прорецензированы членами рабочей группы, которые пришли к вводу, что все замечания и комментарии приняты во внимание, риск систематических ошибок при разработке рекомендаций сведен к минимуму.
Основные рекомендации, уровни доказательности и индикаторы доброкачественной практики приводятся по ходу изложения текста рекомендаций.
Высокотехнологичная помощь больным с мукополисахаридозом типа VI предусматривает наличие подготовленного врача в области диагностики и лечения больных с данным заболеванием, знание им побочных эффектов терапии и методов их коррекции, подготовленного среднего медицинского персонала, возможность выполнения контрольных тестов, технологии вспомогательной терапии.
Определение
Мукополисахаридоз типа VI (МПС VI), известный такж как синдром Марото-Лами, названный так по именам впервые описавших его в 1963 году врачей Марото и Лами, — это лизосомная болезнь накопления, вызванная снижением количества или нарушением функции фермента N- ацетилгалактозамин-4-сульфатазы (арилсульфатазы B (ASB)), которая проявляется в в постепенно усиливающейся задержке роста, приводящей к карликовости, выраженным скелетных деформациях, лицевых дисморфиях, обструкции верхних дыхательных путей, патологии сердечно-сосудистой системы, паренхиматозных органов (гепатоспленомегалия), глаз (помутнение роговицы), слуха (тугоухость). У больных обычно наблюдается нормальный интеллект. По мере прогрессирования заболевания пациенты становятся прикованными к инвалидному креслу вследствие поражения суставов, сердечно-сосудистой патологии, слепоты или компрессии позвоночника. Т.о. заболевание сопровождается снижением качества жизни, и сокращением срока жизни.
Код МКБ-10 (E76.2)
Эпидемиология
Частота возникновения МПС VI составляет от 1 на 238 000 до 1 на 1 300 000. На основании данных о частоте возникновения считается, что в США проживает от 50 до 300 пациентов и около1100больных в развитых странах. Селективный скрининг популяции высокого риска в Бразилии показал, что 19% пациентов с мукополисахаридозом страдали МПС VI. Хотя МПС VI не связывают с определенными этническими группами, регионом эффекта основателя предположительно считают северную Португалию или Бразилию.
Этиология
Ген VI типа мукополисахаридоза (ARSB — arylsulfatase B) картирован на длинном плече хромосомы 5 в участке q11-q13, имеет длину 206 килобаз и содержит 8 экзонов. Ген арилсульфатазы B кодирует одноименный фермент, который осуществляет гидролиз сульфатной группы N-ацетилгалактозамин- 4-сульфата, дерматан сульфата и хондроитин-4-сульфата. Арилсульфатаза В состоит из 492 аминокислот и имеет молекулярный вес 47 kDa. Мутации в гене ARSB ведут к развитию МПСVI.
ДНК-диагностика заболевания показала, что большая часть мутаций у российских больных являются новыми, среди которых чаще встречается мутация R152W (около 1/3 пациентов).
Тип наследования заболевания аутосомно-рецессивный.
Патогенез
МПС VI характеризуется врожденным дефицитом арилсульфатазы В (ASB), одного из пяти ферментов, необходимых для расщепления дерматансульфата — важного компонента соединительной ткани. Дерматансульфат содержится в соединительной ткани разных органов, в том числе кожи, сухожилий, кровеносных сосудов, дыхательных путей и клапанов сердца. Постепенное накопление продуктов распада дерматансульфата в лизосомах всех клеток приводит к необратимым повреждениям клеток и тканей и, соответственно, к дисфункции органов.
МПС VI проявляется только у пациентов с тяжелым дефицитом ферментативной активности арилсульфатазы В (обычно менее 10% от нижней границы нормы). У носителей одного аномального аллеля сохраняется достаточная активность фермента, позволяющая избежать проявлений заболевания.
Классификация
Выделяют две формы болезни – легкую и тяжелую.
Клиническая картина
МПСVI относят к заболеваниям с Гурлер-подобным фенотипом.
Больным с «Гурлер-подобным» фенотипом свойственны внешние специфичные признаки. Обычно они проявляются задержкой роста, диспропорциональным строением скелета (короткие туловище и шея, длинные конечности), грубыми чертами лица, костными деформациями (кифозы, кифосколиозы, воронкообразные деформации грудной клетки), тугоподвижностью крупных и мелких суставов. Наряду с этим отмечаются макроглоссия, полные губы, гипертелоризм глаз, запавшее переносье, дистрофия зубной эмали, кариес, гепатоспленомегалия, пупочные и пахово- мошоночные грыжи, гипертрофия лимфоидного глоточного кольца. Типична патология центральной нервной системы (снижение интеллекта, часто довольно грубое), органов зрения (помутнение роговицы, глаукома), слуха (тугоухость различной степени), а также сердца, сосудов (недостаточность клапанов сердца, гипертрофическая кардиомиопатия) и бронхолегочной системы (апное, синусобронхопатии с образованием обильного количества слизисто-гнойного отделяемого).
Нарушение роста. Синдром Марото-Лами отличают карликовый рост. При быстро прогрессирующих формах заболевания, прогноз роста крайне неоптимистичен: во взрослом возрасте пациенты достигают не более 95 — 100 см в высоту. При медленном развитии заболевания рост может достигать нижней границы нормы и составлять 140 — 150 см.
Аномалии скелета. Клинически аномалии скелета могут быть заметны с рождения — это дорсолюмбарный кифоз в результате передней гипоплазии тел позвонков. Частыми признаками являются кифоз, сколиоз, увеличенный поясничный лордоз и сильные боли в спине Следует учитывать и стараться избежать повреждения спинного мозга в результате нестабильности первого и второго шейных позвонков при дисплазии зубовидного отростка. Кроме того, аномалия формы тел позвонков (уплощение, прогиб назад) могут приводить к сдавлению спинномозговых нервов.
Наблюдаются недоразвитие таза, дисплазия головок бедренных костей и вальгусное положение шейки бедренной кости. Прогрессирующие сгибательные контрактуры пальцев (симптом «когтистой лапы») приводят к снижению подвижности кисти и точной моторики. Сгибательные контрактуры локтевых, бедренных и коленных суставов приводят к снижению общей подвижности и нарушению ходьбы. Способность к ходьбе постепенно снижается и появляется “хождение на пальцах”. В конечном итоге в результате патологии конечностей и позвоночника пациенты могут быть прикованы к инвалидному креслу.
Встречаются аномалии грудной клетки, что негативно сказывается на дыхательной функции.
Термин «множественный дизостоз» используется для описания деформаций скелета пациентов с МПС VI, выявляемых при рентгенографии. Такими изменениями могут быть: заостренные метакарпальные кости, дисплазия головки бедра, дефекты развития тел позвонков, широкие ребра, короткие неровные ключицы.
Аномалии ЛОР-органов и дыхательной системы. Накопление гликозаминогликанов в ротоглотке и дыхательных путях вместе с типичными лицевыми дисморфиями (включая гипоплазию средней части лица и аномалии зубов) приводят к увеличению миндалин и аденоидов, сужению трахеи и бронхов, утолщению надгортанника и голосовых связок, увеличению языка, обструкции верхних дыхательных путей. Как правило, наблюдаются постоянные густые вязкие выделения из носа, хронические риниты.
У пациентов с МПС VI часто наблюдается потеря слуха.
Помимо обструкции дыхательных путей, у пациентов с МПС VI отмечается легочный фиброз. Небольшая и малоподвижная грудная клетка, кифоз, сколиоз и значительный поясничный лордоз являются факторами, предрасполагающими к развитию легочного фиброза.
Обструкция верхних дыхательных путей часто приводит к обструктивному апноэ во сне. Клиническими признаками является ротовое дыхание, храп, апноэ и беспокойный сон. Реже — дневная сонливость, снижение прибавки в весе и отставание в физическом развитии, повышение давления в легких и легочное сердце. В результате беспокойного сна могут наблюдаться трудности с обучением и поведенческие отклонения.
Частые пневмонии могут быть вызваны повышенной секрецией и застоем секрета дыхательных путей.
Аномалии сердца. У пациентов, страдающих МПС VI часто встречаются аномалии сердца, которые иногда становятся причиной летального исхода. Развитие сердечной недостаточности у пациентов, страдающих мукополисахаридозами связано с аномальным накоплением дерматансульфата в сердце и кровеносных сосудах. Наблюдается прогрессирование сердечной недостаточности с возратсом. Симптомы со стороны сердца могут включать следующее. Первым симптомом поражения сердца при МПС VI является прогрессирующая патология клапанов сердца (стеноз и/или недостаточность). Более выражено поражение митрального и аортального клапанов, однако, могут затрагиваться все клапаны сердца. Наиболее часты регургитация на митральном клапане (96%), трехстворчатом клапане (71%) и аортальном клапане (43%). Стеноз митрального и аортальном клапано встречается у 7% пациентов.
У пациентов с МПС VI часто отмечаются аномалии на ЭКГ. Наиболее частые из них: синусовая тахикардия, отклонения электрической оси сердца вправо и влево, увеличение предсердий.
Системная гипертензия может быть сопутствующим заболеванием. Гипертензия может развиваться в связи с сужением аорты или почечной артерии или хронической периодической гипоксией.
Кардиомиопатия при МПС VI редка и не считается характерной для заболевания.
Поражение органа зрения. Нарушение зрения является распространненым и наблюдается у ~40% пациентво с МПС VI, при этом у 15% больных имеется только световосприятие. Большинство пациентов — дальнозоркие.
У пациентов с МПС VI превалирует помутнение роговицы (95%, в 38% из которых нарушение характеризуется как тяжелое). Тяжелое помутнение роговицыи утолщение роговицы может сделать невозможным оценку внутриглазного давления и визуализацию сетчатки и зрительного нерва.
Ретинопатия встречается редко при МПС VI по сравнению с МПС I и II, однако отмечается у некоторых пацеинтов. Могут наблюдаться ночная слепота или ослабление зрения.
У 50% пациентов с МПС VI отмечается отек диска зрительного нерва слабой и средней тяжести, а атрофия зрительного нерва — у 15%. Экскавация диска зрительного нерва связана с повышенным внутриглазным давлением. Отек зрительного нерва может быть связан с повышением внутричерепного давления, накоплением гликозаминогликана в нейронах зрительного нерва, компрессией зрительного нерва или утолщением твердой мозговой оболочки в месте его прохождения.
Повышенное внутриглазное давление часто (50%) встречается у пациентов с МПС VI, однако утолщение роговой оболочки может давать ложно высокие значения. Повышение внутриглазного давления может быть связано с сужением угла передней камеры иридоцилиарной кистой (закрытоугольная глаукома) или отложением гликозаминогликана в трабекулярных клетках с блокированной обратной абсорбцией (открытоугольная глаукома). У некоторых пациентов, у которых ранее отмечалось повышенное внутриглазное давление, оно нормализовалось после пересадки роговицы.
Симптомы, связанные с центральной и периферической нервной системой. Гидроцефалия, поражение спинного мозга и компрессионные нейропатии являются самыми частыми нарушениями со строны нервной системы у пациентов с МПС VI.
У больных документированы такие частые неврологические аномалии, как синдром запястного канала и гидроцефалия, более редки такие нарушения, как цервикальная миелопатия с нестабильностью первого и второго шейных позвонков или компрессией спинного мозга. Обычно пациентам с такими аномалиями диагноз ставится несвоевременно, в следствии чего не вовремя назначается лечение.
Синдром запястного канала (СЗК) вызывается компрессией срединного нерва в результате накопления гликозаминогликанов в связке, удерживающей сухожилия мышц-сгибатаелей. Синдром усугубляется деформацией костей в области запястья. Пациенты с МПС VI редко активно жалуются на характерные боли и парестезии, однако многие из них указывают на симптомы, если им задать прямой вопрос. При электрофизиологических исследованиях у большинства пациентов с МПС VI отмечается прогрессирующий двусторонний СЗК, зачастую в тяжелой форме.
Сообщающаяся гидроцефалия. Считается, что повышенное внутричерепное давление у пациентов с МПС VI вызвано утолщением твердой мозговой оболочки и дисфункцией арахноидальный грануляций. Диганоз сообщающейся гидроцефалии по результатам компьютерной томографии или МРТ часто затруднен, поскольку расширение желудочков может быть связано с атрофией коры головного мозга, поэтому может потребоваться прямое измерение давления в ЦНС. Типичные признаки обструктивного гидроцефалии, такие как головные боли по утрам, рвота и отек зрительного нерва на глазном дне зачастую отсутствуют, хотя у некоторых пациентов может отмечаться резкое ухудшение зрения.
Компрессионная миелопатия. Прогрессирующая компрессионная миелопатия может затрагивать спинной мозг пациентов с МПС VI на разных участках, хотя чаще всего поражается шейный отдел. Причиной этой патологии может быть прогрессирующее накопление гликозаминогликанов в твердой мозговой оболочке и поддерживающих связках, кифосколиоз и костный стеноз. Изначально компрессионная миелопатия может проявляться в виде проводниковых симптомов при неврологическом обследовании, однако позже развивается слабость в нижних конечностях и, впоследствии, спастическая параплегия или квадриплегия.
Диагностика
Врач может заподозрить мукополисахаридоз на основе клинических проявлений. У больного ребенка может наблюдаться сниженная скорость роста, грубые черты лица, деформация скелета, частые инфекции верхних дыхательных путей, увеличение печени и селезенки, скованность в суставах, грубые волосы. Повышение уровня гликозаминогликаналов в моче может указывать на мукополисахаридоз. Выявленный методом тонкослойной хроматографии или электрофореза повышенный уровень дерматансульфата усиливает подозрение на МПС VI, однако могут иметь место ложноположительные и ложноотрицательные результаты. Исследование гликозаминоглаканов в моче применяется для выявления пациентов, подозрительных на МПС VI, и для мониторинга эффективности ферментозамещающей терапии. Образцы низко концентрированной мочи представляют существенную проблему для анализа уровня гликозаминогликанов, и пациенты с МПСVI могут быть пропущены, поскольку дерматансульфат не экскретируется в больших количествах.
Анализ активности фермента является ключевым этапом лабораторной диагностики. Определение активности других сульфатаз рекомендуется для исключения множественной сульфатазной недостаточности, что необходимо сделать, прежде чем начать терапию.
Молекулярно-генетический анализ мутаций в гене ARSB подтверждает диагноз и особо важен для выявления носителей заболевания с последующим их медико-генетическим консультированием и пренатальной диагностикой. Идентификация мутаций начинается с забора образца ДНК пациента, для получения которого используется кровь либо слюна. Большинство мутаций у пациентов с МПСVI могут быть выявлены с помощью стандартного секвенирования (после выделения ДНК используют полимеразную цепную реакцию для того, чтобы амплифицировать кодирующую область гена; каждый полученный ПЦР-продукт затем подвергается секвенированию; последовательность ДНК гена ARSB пациента сравнивают с референсной). Зарубежные исследования показали, что в гене ARSB выявляют миссенс и нонсенс мутации, делеции, инсерции, мутации сайтов сплайсинга. Наиболее частым аллелем является аллель с мутацией c.629A>G (p.Y210C), который находят у 18% пациентов. Кроме того часто встречаются мутации c.1151G>A (p.S384N) – 12,4% пациентов; c.944G>A (p.R315Q) – 11,4% пациентов. Остальные мутации составляют 10% случаев. Некоторые мутации не могут быть обнаружены с помощью секвенирования. Если мутаций не обнаружено в двух аллелях гена ARSB, то рекомендуется провести поиск делеций гена с использованием таких методов как MLPA, ПЦР в реальном времени или серийной сравнительной геномной гибридизации.
Алгоритм диагностики МПСVI представлен на схеме.
Клиническое подозрение на МПСVI: скелетные аномалии и лицевой фенотип → Забор мочи для исследования ГАГ: первая утренняя порция мочи предпочтительна, хранить замороженной → Если выявлен дерматан сульфат и повышен уровень ГАГ перейти к следующему этапу → Забор крови для исследования активности фермента → Выявлена низкая активность фермента ASB Исключен множественный дефицит сульфатаз путем исследования активности других сульфатаз → Молекулярно-генетическое тестирование мутаций в гене ARSB: Идентификация двух патогенных мутаций (по одной от каждого родителя)
Схема. Алгоритм диагностики МПСVI.

Дифференциальный диагноз проводится с системными скелетными дисплазиями и другими лизосомными болезнями накопления.
Так детей с МПСVI от больных со спондилоэпифизарной дисплазией отличает отсутствие характерных для последней рентгенологических изменений трубчатых костей и позвоночника, а также наличие грубых черт лица и патологии внутренних органов (гепатоспленомегалия).
Типичный для МПСVI Гурлер подобный фенотип характерен для ряда болезней накопления. Например, муколипидоз IIB типа имеет внешнее сходство с МПСVI, но при этом заболевании отсутствует повышенная почечная экскреция гликозамингликанов.
Для детей с маннозидозом, в отличие от МПСVI, типичны высокие или средние показатели физического развития, отсутствие тугоподвижности суставов, помутнения роговицы и грубое снижение интеллекта. В моче у этих больных нормальное содержание гликозамингликанов.
Если пациенту поставлен диагноз МПС VI, и у него есть отставание в умственном развитии, то это должно вызывать предположение о множественном дефиците сульфатаз. Следует исключить множественный дефицит сульфатаз путем определения уровня активности других ферментов из группы сульфатаз.
Дифференциальный диагноз следует проводить также с мукополисахаридозами I и II типов. При МПСII наблюдаются типичные узелково-пвпулезные высыпания на коже и отсутствует помутнение роговицы, определяется дефицит фермента идуронатсульфатазы. Для МПСI характерно присутствие в моче фракции гепарансульфата, чего не наблюдается при МПС VI, а также дефицит фермента альфа-L-идуронидазы.
Лечение
Использование ферментозамещающей терапии. Наглазим (galsulfase) – первое специально разработанное ферментозамещающее лекарственное средство, предназначенное и одобренное для лечения больных с VI типом мукополисахаридоза (синдромом Марото-Лами). Производством и распространением препарата занимается фармацевтическая компания BioMarin («Биомарин», США). Наглазим зарегистрирован в США и странах Евросоюза в 2005 и 2006 годах, соответственно. В России препарат зарегистрирован 15.07.2009 года; регистрационный номер: РЛС 005730/01.
Зарубежные клинические исследования препарата продемонстрировали его эффективость и позволили разработать наиболее оптимальную дозу, которая составила 1 мг/кг массы тела [7]. Наглазим вводится внутривенно, медленно (в течение 3-4 часов) с периодичностью 1 раз в неделю, пожизненно. Форма выпуска препарата: флакон 5 мл; 1 мл содержит 1 мг активного вещества.
Подробно приготовление раствора для введения описано в листке- вкладыше к препарату. Наглазим не содержит консервантов и подлежит немедленному ведению. Его нельзя вводить вместе с другими веществами по одной линии. После разведения физиологическим раствором в пакете для инфузий, весь неиспользованный препарат и расходные материалы подлежат утилизации.
Доказано, что Наглазим улучшает у больных с синдромом Марото- Лами показатели тестов 12 минутной ходьбы и трех минутного подъема по лестнице. Так, почти у половины пациентов (48%), получавших введение Наглазима в течение 24 недель, увеличилась длина проходимой за 12 минут дистанции; а 48 недель лечения число таких пациентов повысилось до 64%. Идентичные результаты получены при анализе показателей теста трехминутного подъема по лестнице: у 38% больных улучшились эти показатели после 24 недель лечения, в то время как 54% — после 48 недель. Наряду с этим в процессе лечения зарегистрировано также значительное снижение почечной экскреции гликозаминогликанов.
Перед введением препарата следует оценить состояние дыхательных путей и системы дыхания пациента. Рекомендуется отложить введение Наглазима пациентам с острыми лихорадочными или респираторными заболеваниями. Премедикацию антигистаминными препаратами с жаропонижающими или без них рекомендуется проводить за 30 — 60 минут до начала вливания, чтобы уменьшить вероятность реакций, связанных с введением препарата.
Поскольку у пациентов с МПС VI часто случается апноэ во сне, антигистаминные препараты, вызывающие сонливость, могут повысить риск апноэ. Рекомендуется использовать антигистаминные препараты, которые не обладают седативным эффектом. При введении Наглазима пациентам, которые во время сна получают дополнительный кислород или искусственную вентиляцию легких, эти средства должны быть наготове во время вливания.
Несмотря на премедикацию, в процессе терапии Наглазимом, также как при лечении другими ферментозамещающими препаратами, зарегистрирован ряд побочных реакций. Среди них наиболее частыми являлись: лихорадка, озноб, головная боль, артралгия, рвота, боли в брюшной полости и ушах, кашель, бронхоспазм, остановка дыхания, крапивница, ангионевротический отек, повышение или снижение артериального давления. Характерно также образование антител к препарату.
Рреакции, связанные с вливанием препарата, развиваются у 30 из 55 пациентов, которые получали Наглазим во время клинических исследований. Симптомы обычно исчезали при замедлении или приостановке инфузии, а также введении дополнительных антигистаминных препаратов, жаропонижающих и, иногда, кортикостероидов. Последующие вливания Наглазима выполнялись с меньшей скоростью и сопровождались дополнительным профилактическим введением антигистаминных средств. При возникновении более тяжелых реакций, может потребоваться профилактическое введение кортикостероидов за 12 – 18 часов до введения Наглазима.
Симптоматическая терапия. Больным с синдромом Марото-Лами целесообразно назначение широкого спектра медикаментозных средств: ноотропов, сердечно-сосудистых препаратов, гепатопротекторов, витаминов и медикаментов, направленных на нормализацию минерального обмена с целью предотвращения остеопороза.
Из физиотерапевтических процедур больным с VI типом мукополисахаридоза показаны: электрофорез лидазы на область пораженных суставов, магнитотерапия, парафиновые аппликации и лазерная пунктура.
Широко используются также занятия ЛФК с воздействием на позвоночник и суставы, курсы общего массажа № 10-15 не реже 2-х раз в год.
Обязательна санация хронических очагов инфекции носоглотки и полости рта.
Хирургические операции при синдроме Марото-Лами включают, как правило, грыжесечения, аденотонзиллэктомии и шунтирование гидроцефалии.
Трансплантация гематопоэтических стволовых клеток (ТГСК) при VI типе мукополисахаридоза не проводится.
Особенности проведения анестезии. Наибольшую сложность для анестезиологов представляют дети, страдающие мукополисахаридозом. Наблюдение за осложнениями в этой группе пациентов в Королевском детском госпитале Манчестера показало, что общая частота затруднений интубации составляет 25% в всех подгруппах, а невозможность интубации — 8%.84. Помимо осложнений, связанных с обструкцией дыхательных путей, у пациентов также наблюдаются аномалии лица и черепа, укороченная шея, тугоподвижность в височно-нижнечелюстных суставах, смещенная кпереди гортань, и, у некоторых, нестабильность связки первого и второго шейного позвонков, что требует избегать чрезмерного разгибания шеи. Все это может усложнить ларингоскопию и интубацию. Могут иметь место аномалии сердца, такие как сужение коронарных артерий или легочная гипертензия . Отек легких может усложнить попытки экстубации в послеоперационный период.
Перед проведением любых процедур, связанных с введением седативных препаратов или анестезии, следует оценивать состояние сердца, дыхательной системы и верхних дыхательных путей.
Рекомендуется направить больного на лечение в медицинских центр, где имеется опыт анестезии и ухода за подобными пациентами. Поскольку интубация может представлять проблему из-за аномалий верхних дыхательных путей, рекомендуется привлечь ЛОР-врача для обследования и лечения пациента. Рядом анестезиологов рекомендуется использование метода индукции спонтанного дыхания летучими агентами, ларингеальный масочный воздуховод и оптоволоконную бронхоскопию для проведения интубации.
 Профилактика
Профилактика
Родителям детей с МПС VI рекомендуется консультация генетика, который может пояснить риск рождения ребенка с МПС VI. Если оба родителя являются гетерозиготными носителями МПС VI, для каждой беременности риск рождения ребенка с МПС VI составляет 25%, 50% — риск рождения гетерозиготного носителя, и 25% — вероятность рождения носителя 2 нормальных аллей гена ASB. В семьях, где есть больной ребенок, существует возможность проведения пренатальной диагностики. с этой целью генетик рекомендует родителям соответствующие диагностические лаборатории и медицинские центры.
Программа скрининга новорожденных находится в разработке и может быть доступна уже в обозримом будущем, особенно сейчас, когда разработан метод лечения с помощью ферментозамещающей терапии.
Рекомендуемые обследования
Рекомендованные больным с МПС VI обследования представлены в Таблице 1. По клиническим показаниям частота обследований может быть увеличена. (Таблица 1)
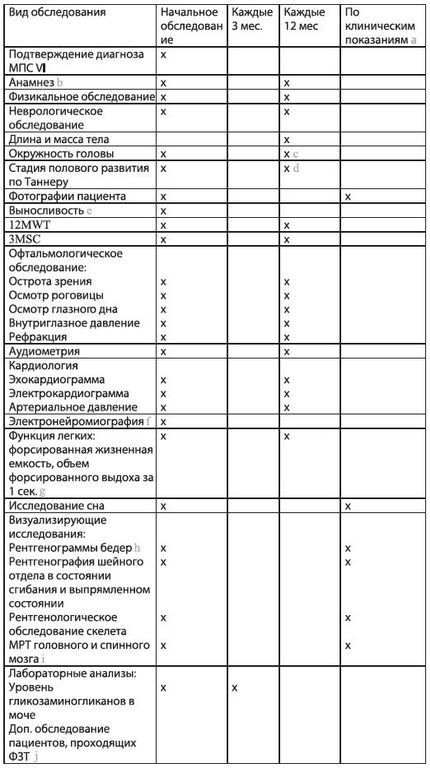 a — «По клиническим показаниям» обычно означает раз в 2 — 3 года, в зависимости от скорости прогрессирования заболевания и клинических симптомов;
a — «По клиническим показаниям» обычно означает раз в 2 — 3 года, в зависимости от скорости прогрессирования заболевания и клинических симптомов;
b — Детей необходимо обследовать чаще;
c Контролировать до остановки роста головы;
d — Продолжать до завершения полового созревания;
e — Парадигма теста на выносливость до и после ФЗТ: расстояние, пройденное за 12 минут (или за 6 минут, но предпочтительным является тот же промежуток времени, что и в предыдущих тестах этого пациента); число ступеней, пройденных вверх за 3 минуты; f Измерение проводимости срединного нерва для контроля синдрома запястного канала;
g — Для проверки функции легких измеряют следующие показатели: форсированная жизненная емкость, объем форсированного выдоха за 1 сек. h Снимок таза в переднезадней проекции и вид сбоку с разведением бедер;
i — МРТ головного и спинного мозга может потребовать общей анестезии, в зависимости от возраста и контактности пациента. Общая анестезия представляет, сама по себе, определенную опасность для пациентов с МПС VI ;
j — Для пациентов, которые проходят курс ФЗТ, измерения следует проводить в начале лечения, на 3, 6, 12, 18 и 24 месяц, а затем — ежегодно.





