

Наследственные заболевания — заболевания, этиологическим фактором которого является мутация. Хромосомные болезни — группа наследственных заболеваний, обусловленных изменением числа и структуры хромосом.
Частота хромосомных болезней составляет — 5-7 на 1000 новорожденных.
Различают следующие хромосомные нарушения:
1). Болезни, связанные с нарушением числа аутосом (анеуплоидии, триплоидии и др.).
2). Болезни, связанные с изменением числа хромосом (трисомиии, моносомии).
3). Болезни, связанные с изменением структуры хромосом (делеции, дупликации, инверсии, транс локации: сбалансированные и несбалансированные).
Наследственные заболевания, обусловленные микроделециями и микродупликациями хромосом (болезни геномного импритинга).
Моногенные болезни — заболевания, обусловленные мутациями на уровне гена.
Тип наследования — аутосомно доминантный, аутосомно-рецессивный, Х-сцепленный доминантный, Х-сцепленный рецессивный.
Митохондриальные заболевания — передаются от матери с митохондриальной ДНК.
Мультифакториальные заболевания, врожденные пороки развития.
В мире описано более 300 наследственных заболеваний с нарушением слуха, различными формами тугоухости, пороками развития ушной раковины.
В статье описаны наиболее часто встречающиеся заболевания, их основные клинические проявления.
Синдром Гольденхара (гемифациальная микросомия, окулоаурикуловертебральная дисплазия) Omim 164210.
Частота развития заболевания — 1:3000 — 1:5000 новорожденных. Этиология заболевания остается до конца неизвестной. Большинство случаев — спорадические. Хотя выявлены семьи, у которых этот синдром передается по аутосомно-рецессивному и аутосомно-доминантному типу наследования. 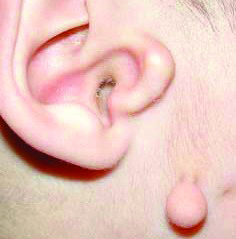 У нескольких детей, имеющих клиническую симптоматику, сходную с Синдромом Гольденхара, были выявлены хромосомные перестройки на 5, 8 и 21 хромосомах. В нескольких спорадических случаях синдрома Гольденхара были выявлены мутации в Гене GSC, который является кандидатным геном для гена HFM. У пяти пациентов — в гене TCOF1, мутации в котором приводят к развитию синдрома Тричера-Коллинза. Основные клинические проявления заболевания: Гипоплазия скуловой или нижниечелюстной области; Макростомия, асимметрия рта (вытянут в оду сторону); Гипоплазия мимических мышц; Микротия; Преаурикулярные выросты, ямки; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Нейросенсорная тугоухость; Эпибульбарные дермоиды; Колобомы верхнего века; Микрофтальм; В 70% случаев поражение одностороннее; Гипоплазия позвонков; Клиновидные позвонки; В некоторых случаях — умственная отсталость, врожденные пороки сердца.
У нескольких детей, имеющих клиническую симптоматику, сходную с Синдромом Гольденхара, были выявлены хромосомные перестройки на 5, 8 и 21 хромосомах. В нескольких спорадических случаях синдрома Гольденхара были выявлены мутации в Гене GSC, который является кандидатным геном для гена HFM. У пяти пациентов — в гене TCOF1, мутации в котором приводят к развитию синдрома Тричера-Коллинза. Основные клинические проявления заболевания: Гипоплазия скуловой или нижниечелюстной области; Макростомия, асимметрия рта (вытянут в оду сторону); Гипоплазия мимических мышц; Микротия; Преаурикулярные выросты, ямки; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Нейросенсорная тугоухость; Эпибульбарные дермоиды; Колобомы верхнего века; Микрофтальм; В 70% случаев поражение одностороннее; Гипоплазия позвонков; Клиновидные позвонки; В некоторых случаях — умственная отсталость, врожденные пороки сердца.
Гемифациальная микросомия с пороками конечностей (141400 Omim).
Тип наследования предположительно аутосомно-доминантный; Большинство случаев — спорадические; Врожденные пороки конечностей; Трехфаланговые пальцы; Удвоение фаланг; Расщелина мягкого и твердого неба; Гемифациальная микросомия; Преаурикулярные выросты; Кожные выросты в области нижней челюсти; Микротия; Атрезия наружного слухового прохода; Кондуктивная тугоухость; Данный синдром может рассматриваться как вариант Синдрома Гольденхара.
Бранхио-ото-ренальный синдром (Синдром Менльника-Фрейзера)(Omim 113650).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1, расположенного на длинном плече хромосомы 8 (8q13.3); Частота заболевания 1:40000 новорожденных; Длинное лицо; У 10 % пациентов -парез лицевого нерва; Нейросенсорная тугоухость (20%); Кондуктивная тугоухость (30%); Смешанная тугоухость (50%); Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Атрезия или сужение слезного канала; Дисплазия Мондини; Прогнатия; Высокое небо; Расщелины твердого и мягкого неба; Расщепление небного язычка; Жаберные щели, расположенные симметрично с обеих сторон — редко; Дисплазия почек; Аплазия почек; Поликистоз почек; Пороки собирательной системы почек; Врожденный вывих бедра; Незавершенный поворот кишечника.
Бранхио-отический синдром (Omim 602588).
Тип наследования — аутосомно-доминантный; Заболевание обусловлено мутациями в гене EYA1; Нейросенсорная тугоухость; Кондуктивная тугоухость; Смешанная тугоухость; Микротия; Чашеобразные уши; Деформация мочки, наружного уха; Бранхиогенные кисты или свищи; Стеной наружного слухового прохода; Пороки развития среднего и внутреннего уха; Данный синдром иногда рассматривают, как вариант синдрома Мельника -Фрезера, но большинство исследователей склоняются к тому, что это разные заболевания, вызванные аллельными мутациями гена EYA1.
Бранхио-окуло-фациальный синдром (Omim 113620).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TFAP2A, расположенного на коротком плече 6 хромосомы — 6р24.3. Основные клинические проявления заболевания: Пренатальная задержка роста; Задержка роста; Бронхиогенные кисты или свищи; Атрофия или очаговая аплазия кожи; Гемангиомы на коже; Атрезия слезного протока; Колобомы; Микрофтальм; Анофтальмия; Гамартома сетчатки; Дермоидные кисты глазницы; Эпителиальные кисты радужной оболочки; Катаркта; Миопия; Монголоидный разрез глаз; Телекант; Миопия; Низкопосаженные, ротированнын ушные раковины; Гипоплазия верхней части завитка; Микротия; Преаурикулярные выросты; Кондуктивная тугоухость; Аномалии среднего уха; Свищи над ушами; Широкое, запавшее переносье; Псвевдо-расщелины, расщелины губ, неба; Аномалии зубов; Микрогнатия; Гипоплазия скуловых костей; Агенезия почек; Дисплазия почек; Клинодактилия, полидактилия, грубое нарушение дерматоглифики; Гипоплазия ногтей; Эктопия тимуса; Ранняя седина; Умственная отсталость легкой степни; Гнусавый голос.
Синдром микротии нарушения слуха и расщелины неба (Omim 612290).
Тип наследования: аутосомно-доминантный, аутосомно-рецессивный. Заболевание обусловлено мутациями в гене HOXA2, расположенном на коротком плече 7 хромосомы — 7р15.2. Основные клинические проявления заболевания: Микротия; Сужение наружного слухового прохода (выраженное — при аутосомно-рецессивных формах); Деформация слуховых косточек; Заполнение барабанной полости костной тканью; Сращение слуховых косточек (при аутосомно-рецессивных формах); Кондуктивная тугоухость; Полная потеря слуха (при аутосомно-рецессивных формах); Расщелина твердого и мягкого неба.
Синдром Таунза-Брукса (Omim 107408).
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене SALL1, который расположен на длинном плече 16 хромосомы 16q12.1. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Смешенная тугоухость; Маленькие, чашеобразные уши со свисающим завитком; Преаурикулярные выросты; Трехфаланговый, раздвоеннный широкий 1 палец; Преаксиальная полидактилия; Псевдоэпифиз 2-ой пястной кост; Сращение костей кистей и стоп; Гипоплазия или аплазия 3 пальцев; Клинодактилия 5 пальцев; Эктопия или атрезия заднего прохода; Врожденные пороки влагалища и прямой кишки; Агенезия или гипоплазия почек, поликистоз почек; Пузырно-мочеточниковый рефлюкс; Расщепление мошонки; Гипоспадия; Иногда — умственная отсталость.
Синдром Фрейзера (синдром Фрайзера) (Omim 219000).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в генах FRAS1, расположенного на длинном плече 4 хромосомы — 4q21.21, GRIP1, расположенного на длинном плече 12 хромосомы — 12q14.3, и гена FREM2, расположенного на длинном плече 13 хромосомы — 13q13.3. Основные клинические проявления заболевания: Двусторонний криптофтальм; Недоразвитие глазного яблока; Низкая граница роста волос на лбу на стороне поражения; Западение лобной кости на стороне поражения; Широкий нос; Запавшее переносье; Недоразвитие крыльев носа; Чашеобразные ушные раковины; Микроотия; Атрезия слухового прохода; Пороки среднего уха; Кондуктивная тугоухость; Расщелина твердого и мягкого неба; Стеноз трахеи; Атрезия трахеи; Кожная синдактилия на кистях и стопах; Крипоизм; Гипоспадия; Атрезия влагалища; Агенезия почек; Микроцефалия; Менингоцеле; Энцефалоцеле; Более 20% детей погибают в первый год жизни.
Синдром Тричера-Коллинза (Синдром Франческетти, челюстно-лицевой дизостоз) (Omim 154500).
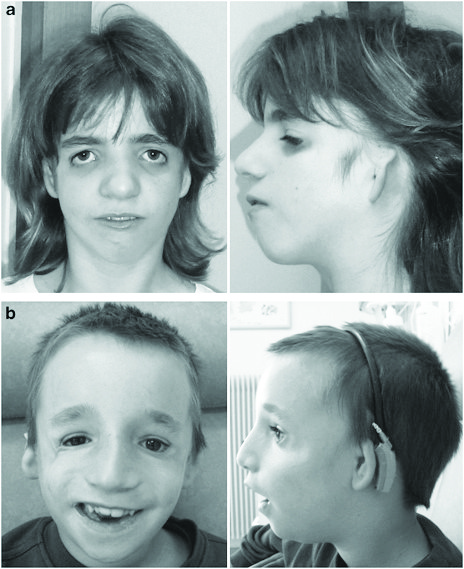 Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TCQF1, расположенного на длинном плече 5 хромосомы — 5q31.3-q32. В настоящее время три типа данного заболевания. Основные клинические проявления заболевания: Антимонголоидный разрез глаз; Гипоплазия скуловых костей; Микрогнатия; Колобома нижнего века; Отсутствие ресниц на нижнем веке; Аномалии ушных раковин; ВПР наружного слухового прохода; Кондуктивная тугоухость; Расщелина неба; Нарушения глотания; Низкая граница роста волос на висках; Гипоплазия средней трети лица; Врожденные пороки сердца; Крипторхизм.
Тип наследования — аутосомно-доминантный. Заболевание обусловлено мутациями в гене TCQF1, расположенного на длинном плече 5 хромосомы — 5q31.3-q32. В настоящее время три типа данного заболевания. Основные клинические проявления заболевания: Антимонголоидный разрез глаз; Гипоплазия скуловых костей; Микрогнатия; Колобома нижнего века; Отсутствие ресниц на нижнем веке; Аномалии ушных раковин; ВПР наружного слухового прохода; Кондуктивная тугоухость; Расщелина неба; Нарушения глотания; Низкая граница роста волос на висках; Гипоплазия средней трети лица; Врожденные пороки сердца; Крипторхизм.
Синдром Пендреда (Omim 274600).
Тип наследования — аутосомно-рецессивный. Заболевание обусловлено мутациями в гене SLC26A4, расположенном на длинном плече 7 хромосомы, 7 q22.3. Основные клинические проявления заболевания: Нейросенсорная тугоухость; Аномалии развития структур костного лабиринта внутреннего уха; Неполное разделение завитка улитки; Дисплазии лабиринта улитки; Широкий водопровод преддверия; В подростковом возрасте — увеличение размеров щитовидной железы; Гипотиреоз; Карцинома щитовидной железы.
Синдром Варденбурга.
Тип наследования для I, II и III типов — аутосомно-доминантный, для четвертого — аутосомно-рецессивный. Основные клинические проявления заболевания: 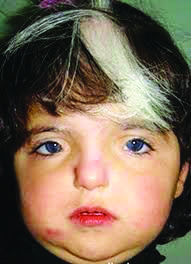 Короткие глазные щели; Телекант; Нос прямой; Переносье широкое, высокое; Гипоплазированные крылья носа; Густые брови; Синофриз; Очаги депигментации на коже; Бесцветные ресницы и брови; На глазном дне — картина, характерная для альбинизма; Седая прядь волос на лбу; Аплазия заднего полукружного канала; Нейросенсорная тугоухость; Выступающая нижняя челюсть; Высокое стояние лопаток; Добавочные ребра; Spina bifida; Врожденные пороки сердца.
Короткие глазные щели; Телекант; Нос прямой; Переносье широкое, высокое; Гипоплазированные крылья носа; Густые брови; Синофриз; Очаги депигментации на коже; Бесцветные ресницы и брови; На глазном дне — картина, характерная для альбинизма; Седая прядь волос на лбу; Аплазия заднего полукружного канала; Нейросенсорная тугоухость; Выступающая нижняя челюсть; Высокое стояние лопаток; Добавочные ребра; Spina bifida; Врожденные пороки сердца.
В заключение хотелось бы сказать, что при подозрении у ребенка на наследственную патологию, сопровождающуюся различными формами нарушения слуха, ребенок должен получить консультацию врача-генетика, и при необходимости ему будет проведено молекулярно-генетическое обследование, исследование кариотипа для установления точного диагноза наследственного заболевания, уточнения прогноза развития заболевания и возможности медико-генетического консультирования семьи.





